
Staphylococcus aureus and its antibiotic-resistant strains are the cause of soft tissue infections representing some severe life-threatening infections. These situations have caused great concern for its treatment worldwide. Thus, the need to introduce new antibiotics or an alternative to antibiotics markedly increasing. Antimicrobial peptides (AMPs) have been shown to have various properties and uses in the biological system since their discovery. This review is based on the increasing concern for S. aureus, its resistant strains, the associated infections, pathogenicity, and the mechanism of resistance to antibiotics. Lastly, the overall significance of AMPs against S. aureus showed that they can be ideal candidates as an alternative to antibiotics with high potential for future therapeutics.
Bacterial infection by antibiotic-resistant bacteria has been a serious global public health concern in the last decades. Usually, antibiotics exhibit good activity against bacterial infections and all of them come along with side effects. However, the emergence of antibiotic-resistant strains with time has further worsened the condition with increasing pressure on the clinical setting [1]. These have increased the morbidity and mortality rate and prolonged hospital stay [2, 3, 4]. These antibiotic-resistant strains are the leading cause of hospital and community-acquired infections. The overuse, misuse of antibiotics, and an incomplete course of antibiotics are among the main reasons for antibiotic resistance [5, 6, 7]. Further, the ability of the bacteria to evolve and gain resistance to antibiotics via various modalities like reduced permeability, efflux via pumps, alterations of the target enzyme, horizontal gene transfer, and ability to produce enzymes which degrade the antibiotic, has made the situation more critical [8]. Thus, this scenario has pushed the researcher and clinical settings to develop new antibiotics or generate some new alternatives to antibiotics.
The bacterial strains that are normally acquired even inside the intensive care units (ICU) are Acinetobacter baumannii (19.5%), Enterococcus spp. (10.6%), Staphylococcus aureus (10.7%), Pseudomonas aeruginosa (15.6%), Stenotrophomonas maltophilia (11.5%), and Klebsiella pneumoniae (9.7%) [9] and the infections caused by these antibiotic-resistant bacteria are hard to cure. According to the World Health Organization (WHO) report of 2017 regarding antibiotic resistance, they listed resistant bacteria based on three priority levels as critical, medium, and high. Where S. aureus, vancomycin-resistant S. aureus (VRSA) and methicillin-resistant S. aureus (MRSA) were listed as a high priority [1]. However, in 2019, the Centre for Disease Control and Prevention (CDC) listed bacteria and fungi as urgent, serious, and concerning. According to their report, MRSA is listed as a serious threat [10]. Moreover, the current uprising of multi-drug resistant strains has made the situation even more critical which calls for modalities to overcome bacterial antibiotic resistance [12, 13]. Thus the interest to develop new antibiotics with proper use is highly increasing. In this respect, the development of an alternative to antibiotics is inevitable. The application of nanoparticles, combined therapy (conjugate formations), plant extracts, algal metabolites, and antimicrobial peptides are some of the common alternatives.
Antimicrobial peptides (AMPs) are amphipathic and their natural occurrence provides innate immunity via non-specific and effective defensive activity against invading pathogens. Further, their ability to enter bacteria, mostly soluble in water or aqueous buffer or organic solvent, possibility to modify and their ability to form a secondary structure like beta sheets and helices, has made them a promising candidate alternative to antibiotics [4, 14, 15]. AMPs showed prominent activity against fungi and bacteria with decreased resistance compared to antibiotics. AMPs also display anticancer activity. They have limitations including stability, non-specific interaction, high production costs, lack of information regarding their mechanism of action, and sometimes high toxicity [16]. Despite these drawbacks, AMPs are also clinically tested and approved for their appearance in the world of medicines.
The Staphylococcus aureus and its related resistant taxonomic classes can sustain harsh conditions like human skin. Fig. 1 gives a clear view of the timeline for the emergence of S. aureus resistant strains. They are opportunistic pathogens which can cause opportunistic infections. They are the cause of both community and healthcare-associated bloodstream infections. They cause infections via various production of secreted and multiple cell surface virulence factors. Further, S. aureus endovascular pathogenesis is the result of the cumulative effects of different virulence factors [17]. The presence of protein A (Spa) on the S. aureus surface acts as a virulence factor that prevents the immune system from performing phagocytosis [18, 19]. Moreover, they are associated with soft tissue infections like surgical site infections, folliculitis, cutaneous abscesses, boils, carbuncles, impetigo, scalded skin syndrome, purulent cellulitis, and furuncles. They are also responsible for life-threatening infections like sepsis, bacteremia, pneumonia, toxic shock syndrome, osteomyelitis, pseudomembranous enteritis, and endocarditis [20]. S. aureus can colonize in the region of lesion of atopic dermatitis patients as well as patients with chronic infections (up to 93% approximately) like chronic leg ulcers. However, S. aureus also plays a role in sepsis in burn patients leading to death [21].
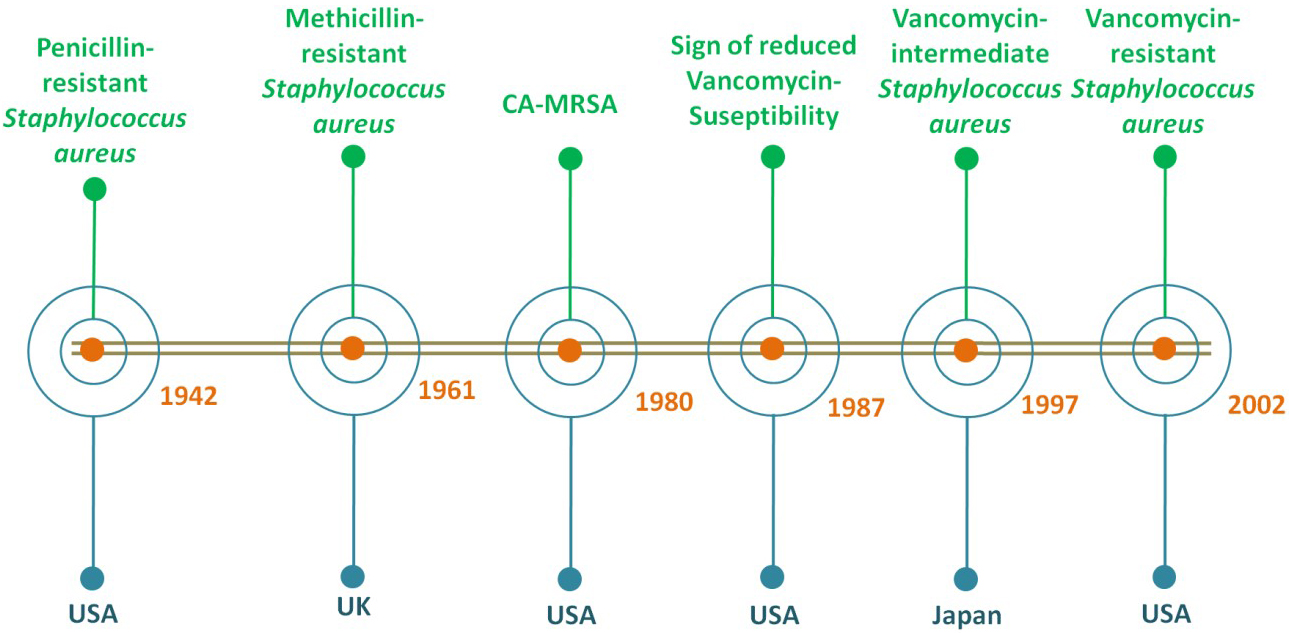 Fig. 1.
Fig. 1.The timeline based on the first reports of the emergence of S. aureus resistant strains since 1900s.
MRSA, a significant nosocomial pathogen with versatile nature associated
with postsurgical wound infections in hospitals. The first case was in the United
Kingdom in the year 1961 [22]. Reports by the WHO showed that out of most cases
of MRSA infection, 64% of people die in comparison to people with non-MRSA
infections [23]. They may be transmitted via physical contact but rarely by air.
The infection by MRSA has increased morbidity, hospital stays, mortality, and
overall cost. Reports showed that S. aureus contains at least 11 major
types (I-XI) of the staphylococcal cassette chromosome mec
(SCCmec) mobile elements that are responsible for the emergence of MRSA
[24]. Therefore, the overall frequencies of the healthcare-associated (HA) and
community-associated (CA) MRSA infection and their ability to gain antibiotic
resistance have increased [25]. Furthermore, the presence of a hypervariable
region in the spa gene (X
The first case of a CA-MRSA infection was reported in the USA in the year 1980. Moreover, significant epidemiological changes in the CA-MRSA had been observed in the past decade [26]. Infections caused by CA-MRSA are usually skin infections but rarely necrotising pneumonia with a high tendency to infect young individuals living in the community. Patients with necrotizing pneumonia often seem to have influenza-like sickness or influenza virus infections with the symptoms of multiple cavity lung infiltrates leads to a mortality rate of more than 50% [27]. They also cause severe infections like Waterhouse-Friderichsen syndrome, necrotising fasciitis, and sepsis. CA-MRSA has a distinct pattern of resistance containing SCCmec IV or V or VI with resistance toward fewer categories of antibiotics [28, 29, 30]. Reports also showed that most CA-MRSA produces a bi-component cytolytic toxin for neutrophils encoded by lukSF-PV genes [31, 32].
The isolates of CA-MRSA mostly contain a phage-encoded Panton-Valentine
leukocidin (PVL). PVL is a toxin that is associated with severe infections like
necrotising pneumonia and also skin infections [33]. It causes necrosis of
epithelial cells and human leukocytes lysis. They also produce an
However, other dangerous isolates are the HA-MRSA that can survive in the hospital environment due to their multidrug resistance ability. The HA-MRSA can colonize in patients who have been admitted to the hospital and underwent medical treatments therein or were recently discharged from the hospital. Even more than six months, the carrier state can persist in these patients. Thus, the patient infections caused by MRSA within the healthcare system or in hospitals are known as healthcare-associated MRSA infections [35]. Albeit, the bacterial peptides group named the phenol soluble modulins (PSM) which causes inflammation, skin infection, and neutrophil cytolysis, is found mainly in CA-MRAS. PSM showed lower expression in HA-MRSA isolates [36]. HA-MRSAs are resistant to a greater number of antibiotic categories due to the presence of SCCmec I, II, and III. These elements facilitate the survival of these pathogens in the healthcare system [37]. Further, the horizontal gene transfer of SSCmec III among S. aureus is not feasible due to the larger size of the mobile element. Thus, the possible spread of HA-MRSA in the community is through healthcare workers being the carriers which come in contact with the community [38, 39, 40]. Thus, more precaution measures are needed to be taken by the healthcare workers.
Antibiotic resistance in S. aureus against vancomycin being the
last resort antibiotic against infections caused by gram-positive bacteria is
also a major concern [4]. According to the Clinical and Laboratory Standard
Institute depending on the susceptibility to vancomycin, S. aureus is
classified into 3 groups including VRSA with MIC
The VISA was discovered in 1997 in Japan. However, retrospective studies
regarding vancomycin susceptibility were observed around 1987 in the USA. They
are partially resistant to vancomycin due to polygenic factors. It includes a
series of mutations in genes which encode molecules that are related to cell
envelope biosynthesis, cell wall thickness increment, altered surface protein
profile, changes in growth characteristics, and other factors [42, 43, 44, 45]. Though it
is accepted that VISA occurred due to the accumulation of a series of gene
mutations, only a few are important like genes encoding two-component regulatory
systems (GraSR, WalKR, and VraSR) [46, 47, 48, 49, 50, 51]. However, the detailed molecular
mechanism of VISA resistance is yet needed to be explored by researchers. The
VISA infections are related to persistent infections, hospitalization, prolonged
vancomycin treatment or failure, and lack in clinical settings. Moreover, the
heterogeneous VISA (hVISA) is the S. aureus population with vancomycin
MIC ranges from
Vancomycin-resistant enterococci (VRE) were identified in Europe which was found endemic in ICUs soon after its discovery [54, 55]. The resistance mediated by transposon Tn1546 present in a conjugative plasmid caused an increased risk in the spread of vancomycin resistance to universally susceptible microorganisms including S. aureus [56, 57, 58, 59]. Further, it was confirmed that in a mixed infected mouse, the van elements were transferred to MRSA from Enterococcus faecalis but it did not spread hence causing a lesser concern [60]. However, there are cases of co-colonization and co-infection of VRE and MRSA, which are common in patients with prolonged hospital stay as well as stay in ICU [61, 62]. The first VRSA isolate was reported in 2002 from Michigan, USA [63, 64]. In numerous cases, isolates of both VRE and VRSA were obtained together from the same patients [65]. It has also been reported that vancomycin acts as a selective pressure for its resistance imparted by vanA gene clusters. The majority of hospital-associated infections caused by VRSA isolates belong to clonal complex 5 (CC5) lineages [66, 67]. Therefore, the misuse of vancomycin for treatment has to stop. Whereas, the development of alternatives is increasingly required.
The resistance towards different antibiotics in S. aureus has emerged due to various mechanisms such as acquiring mobile genetic elements via horizontal gene transfer, increasing the expression of efflux pumps and mutations altering target binding sites [68]. Fig. 2 provides the overall mechanisms of antibiotic resistance in S. aureus.
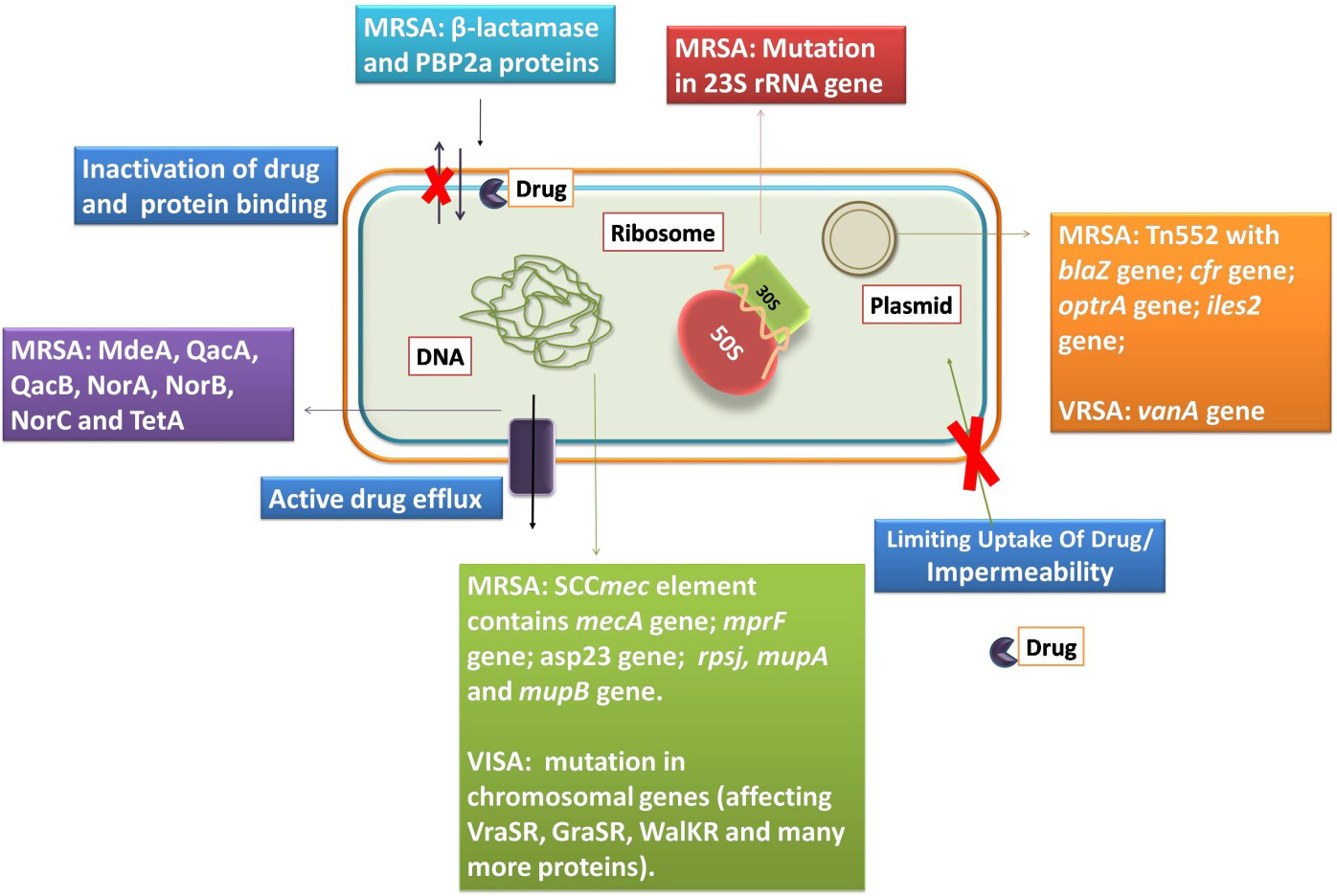 Fig. 2.
Fig. 2.The S. aureus and its mechanisms of resistance to antibiotics.
The first reported penicillin-resistant S. aureus was described
in 1942, in the USA [69]. The production of
MRSA strains contain a large mobile genetic element named SCCmec containing the mecA gene responsible for resistance. Thus, resistance to methicillin and oxacillin is mediated via the acquisition of this mecA gene (encodes PBP2a). PBP2a is not interacting with drugs due to the presence of the active site serine (i.e., of the transpeptidase) in a deep pocket [72, 73]. According to reports, CA-MRSA carries a small SCCmec with higher virulence but lower resistance to multiple antibiotics [74, 75]. The mecA gene is expressed upon exposure to a drug that is under the regulation of MecIR regulatory protein [72]. However, in the presence of BlaI and BlaR, the activity of mecA gene expression was seen to be repressed. Thus, the presence of Mec and Bla regulator can cause a variation in the PBP2a expression.
Further, the non-synonymous or nonsense mutation in mecA can usually cause a change in the amino acid sequence of PBP2a resulting in ceftaroline resistance [76, 77]. It was observed that when passaged in ceftaroline the COL strain showed a mutation in PBP2, PBP4, and gdpP with a high level of resistance towards it [79]. It was later reported the wild type MRSA also showed ceftaroline resistance [78]. Hence, prolonged treatment with ceftaroline can lead to the emergence of resistance to this antibiotic.
MRSA infections like endocarditis and bacteremia were treated with daptomycin, an alternative to vancomycin which was approved in 2003 in the USA. However, certain. aureus strains showed some changes like enhanced membrane fluidity, lower daptomycin surface binding, changes in the cytoplasmic membrane, increase net positive charge, and reduced susceptibility to daptomycin-induced depolarization [80]. When the gene mprF encoding for the lysyl-phosphatidyl glycerol synthetase got mutated, it increased the outer membrane positive charge and reduced the susceptibility to daptomycin [81]. The inactivation of the asp23 gene causes decrease in daptomycin susceptibility [82]. Thus, these alterations in S. aureus strains facilitated the acquisition of resistance to daptomycin.
Furthermore, MRSA also acquired resistance to oxazolidinone via a
mutation in the 23S rRNA, to chloramphenicol-florfenicol resistance
(cfr) gene, and optrA gene (influx or efflux of drugs) [83, 84]. Aminoglycoside modifying enzymes like bidomain AAC(6
Vancomycin was antibiotic of choice for the treatment of MRSA in the late 1980s. However, vancomycin resistance emerged in the same year in VRE. Later, it was followed by reports of vancomycin resistance in S. aureus.
The mechanism of action of vancomycin is to bind the D-Ala-D-Ala dipeptide of lipid II. This binding prevents the transpeptidation and transglycosylation in gram-positive bacteria [95]. In S. aureus, the cell wall is underneath the polysaccharide capsule layer that allows host-pathogen interactions and helps to maintain cell integrity [96]. It consists of N-acetylmuramic acid cross-linked to N-acetylglucosamine by stem pentapeptides (UDP-Mur-NAc-L-Ala-D-iso-Gln-L-Lys-D-Ala-D-Ala) and glycine bridges. Each precursor is synthesized in the cytoplasm of the cells and transported to the cell wall division septum for assembly [97]. Vancomycin resistance in VRSA is due to the presence of the vanA operon. It is present in the Tn3 family of the mobile transposable element named Tn1546. The latter is a part of both conjugative and non-conjugative plasmids originally present in VRE [98]. These conjugative plasmids or transposons were transferred to S. aureus from VRE by horizontal gene transfer thereby establishing VRSA.
The vanA operon-mediated resistance depends on two main events. The first is the synthesis of the D-Ala-D-Lactate peptidoglycan precursor that does not bind to vancomycin. The second, the hydrolysis of the dipeptide precursor (D-Ala-D-Ala) that binds to vancomycin [99]. The vanA operon is composed of a minimum of 7 genes (vanRSHAXYZ). It includes two promoters for each ORF (open reading frames). Whereas the regulatory apparatus is encoded with the help of vanS (membrane-bound histidine kinase) as well as vanR (cytoplasmic transcriptional regulator) representing the two-component system that regulates the transcriptional activation of all vanHAXYZ genes [100]. The VanS (sensor kinase) is anchored to the cytoplasmic membrane with the help of two transmembrane segments. This anchoring helps to predict the sensory domain. It also binds to vancomycin with the help of its ligand-recognition domain and ATP-dependent autophosphorylation (i.e., present on the highly conserved region of histidine residues) [101]. The transfer of the phosphoryl group to the VanR (the response regulator) facilitates its dimerization, increasing its DNA binding affinity, and leads to transcriptional activation of both promoters in the vanA gene cluster. Besides the regulatory genes, the vanA gene cluster also includes the vanH gene which encodes for the dehydrogenase that reduces pyruvate to D-Lactate; vanX encodes for a D,D-dipeptidase that hydrolyzes the dipeptide D-Ala-D-Ala, preventing its incorporation in LIPID II precursors; whereas, vanA which encodes for the ligase is responsible for the synthesis of the dipeptide D-Ala-D-Lac dipeptide. The vanY gene encodes for D,D-carboxypeptidase which helps to eliminate the natural peptidoglycan precursors. The role of the vanZ gene in antibiotic resistance is not known yet but it is usually referred to have an accessory function [102]. Thus, the replacement of D-Ala-D-Ala by D-Ala-D-Lactate helps to prevent vancomycin binding and confers resistance to this antibiotic.
In the case of VISA, isolates gain resistance by obtaining multiple mutations in chromosomal genes due to treatment failure by vancomycin or during prolonged antibiotic treatment. This affects cell homeostasis and cell wall biosynthesis like reduction in crosslinking providing false D-Ala-D-Ala for binding and increase thickness [52, 103]. Due to these mutations, the hVISA and VISA are gaining increased resistance. The VISA strain Mu50 showed a mutation in MsrR which is responsible for the addition of secondary wall teichoic acid and wall polymer capsular polysaccharide to peptidoglycan. Another mutation affects the VraSR, a two-component signal transduction system (TCSTS). This mutation not only caused the constitutive expression of the VraR (response regulator) but also increased the expression of more than 40 genes which are related to cell wall biosynthesis and its stimulation. Thus, these mutations give rise to hVISA features of this strain.
Similarly, mutations in graR (response regulator of GraSR TCSTS) and RNA polymerase B subunit led to the VISA phenotype. This characteristic VISA phenotype emerges upon exposure to vancomycin. Furthermore, the mutation in sle1 and fdh2 reduced autolytic activity, creating a strain with indistinguishable Mu50 phenotype [104]. In strain Mu3, mutations in rpoB, walK, rpoC, cmk, and other genes were the key reason for the emergence of VISA strains [105]. Similarly, in the ST239 VISA strain a mutation in WalKR (a TCSTS) was crucial for some VISA strains [106]. Due to mutations in these TCSTS, dtl, and rpoc, the VISA strain also displayed resistance to daptomycin as they are also the pathway that overlaps with daptomycin resistance in other strains [107]. Thus, the mutation due to prolonged and failed treatment as well as gene acquisition are the reason for the appearance of these resistant strains. Thus, the burning need for new antibiotics or alternatives to antibiotics is inevitable.
Alexander Fleming first discovered the lysozyme, an enzyme which displayed antimicrobial activity which was obtained from the human nasal mucus and tears in1922 [108]. Lysozymes are antimicrobial enzymes with AMPs like properties that are widely present in animals. However, in 1939, the first reported antimicrobial peptides named Gramicidin A, B, and C, was used for clinical purposes [109]. AMPs usually consist of 10-50 amino acid residues with characteristics like net positive charge, presence of secondary structure, and amphipathic nature. They are part of the host defense of almost all living organisms. They not only help in killing microbes but also control the host physiological functions [110]. In the past decade, the number of studies increasing our understanding of the antibacterial activity of AMPs has considerably increased. We have used parameters like antimicrobial peptides against Staphylococcus aureus, antibacterial, and antimicrobial to place a search in the Web of Science portal. The results showed the total number of articles was 1467 from the beginning of 2000. There is a steady increase in interest in this field of AMPs until 2020, as shown in Fig. 3. It is well known that a wide variety of AMPs are present in each species [111], and it is beyond the scope of any review to give an overview of all of these peptides. Thus, a complete directory of the AMPs is not possible. As such, we also did not intend to do so in this review. However, information about the S. aureus, associated infections, and mechanisms of resistance to highlight the key targets and threats associated is presented herein. Further, the key functions of AMPs and their recent activity are demonstrated that elevate their importance and level of consideration for their study, development, and clinical value.
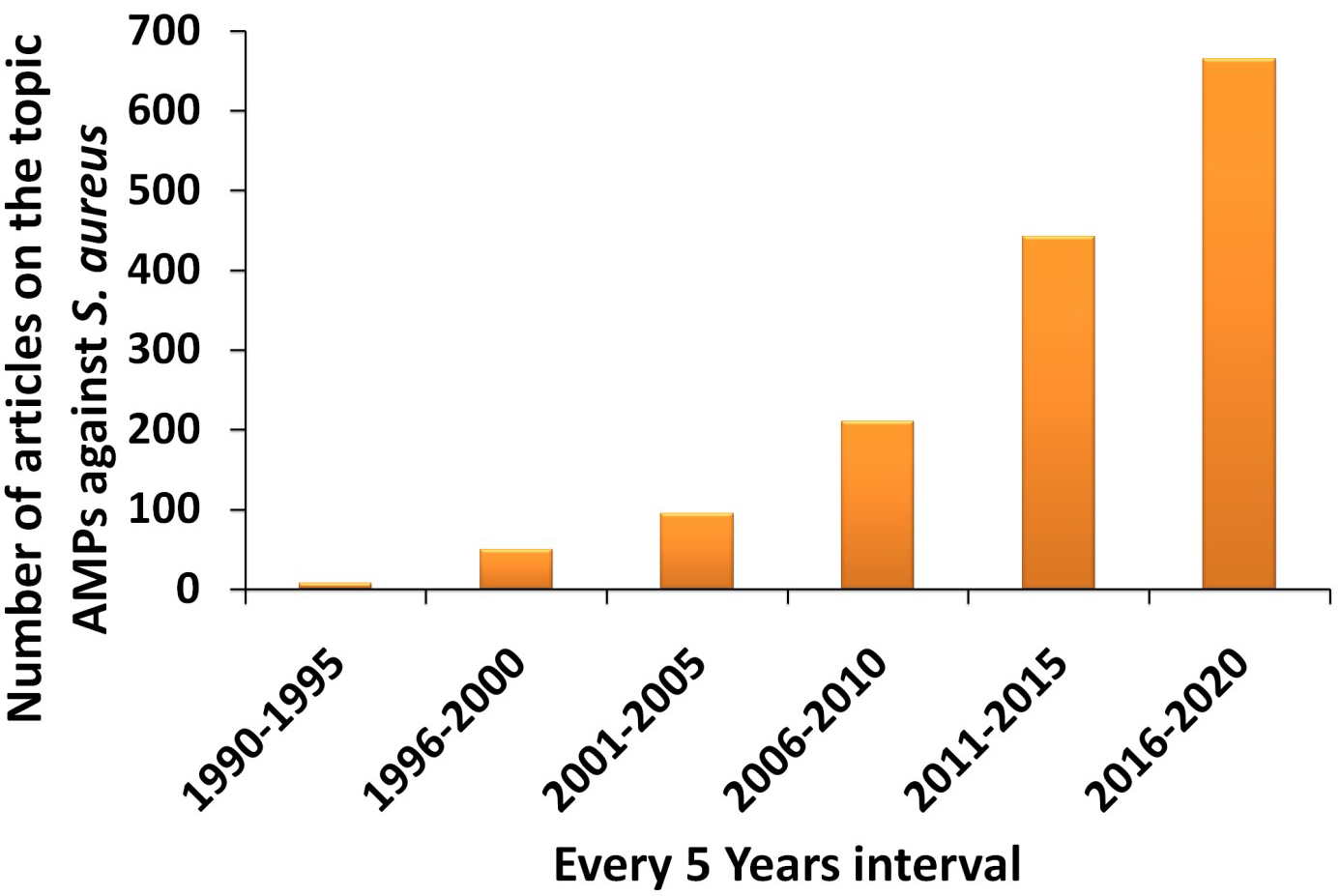 Fig. 3.
Fig. 3.Statistical chart showing the increase number of articles on AMPs against S. aureus from year 2000 until 2020.
Initially, the innate immune system provides the first line of defense against pathogenic invasion and colonization. It consists of physical and chemical defense attributes. The genes encode for host defense peptides as part of the host biological defense; these peptides are also known as antimicrobial peptides [112, 113]. Studies consistently revealed that in response to infections, cationic peptides (like defensins) were produced by phagocytic cells [114]. A human liver-specific peptide is known as hepcidin. Hepcidin is a cysteine-rich peptide that makes its similar to defensins. They have an iron-regulatory activity as well as antimicrobial activity [115]. Reports showed that AMPs from higher organisms have broad-spectrum activities against microorganisms [116]. The expression of genes that encode for the AMPs in mammals is not tissue-specific but occurs throughout the body. Although, reports showed that a combination of host defense molecules is produced at a single site by the expression of multiple AMP due to the coordinated transcriptional regulation of AMP genes [117, 118].
Cathelicidins and defensins are the most abundant human AMPs. The
The importance of the host-derived peptides is not only restricted to
antimicrobial activity during microbial invasion or phagocytosis. However, they
can also cross-talk with adaptive and innate immunity. It was reported that
Numerous approaches like computational design approaches, site-directed
mutagenesis, template-assisted methodologies, mechanism-based strategies, and
synthetic libraries are in use to design AMPs. Although the strategies are
different conceptually, the aim is identical. All these strategies attempt to
develop de novo designed synthetic peptides or modify the naturally
occurring peptides for improved antibacterial activity [134]. Furthermore, to
understand AMPs activity at different pH values, solubility, structure,
composition, molecular weight, charge, amphiphilicity, and other parameters,
different biophysical and biochemical methods are used [135, 136]. According to
the secondary structures, peptides are divided into four groups named as
Following the characterization, the biological activity of the peptides needs to be evaluated. The antibacterial activities of the peptides against the bacterial strains are evaluated by determining the minimal inhibitory concentrations, and minimum bactericidal concentration, growth curve analysis, disk diffusion assay, live dead cell assay and other assays [140, 141]. However, after obtaining the information about the activity, an in depth understanding of the mechanism of action (which can vary between peptides) is required.
The actual mechanism of action of most AMPs is still not known in detail. The most general mechanism of action is the cytoplasmic membrane pore formation that includes a carpet-like model, barrel-stave model, and toroidal model. The carpet-like model is based on the AMPs concentration. When the AMPs concentration is high, they become accumulated on the cell surface and dissolve in the cell membrane [142]. In the barrel-stave model, the formation of the membrane-spanning pores occurs due to the direct interaction of AMPs into the target membrane [143]. Lastly, in the toroidal model, a curvature is induced in the membrane due to membrane-spanning pores by AMPs with intercalated lipids [144]. Additionally, other mechanisms by which peptides act are charged lipid clustering, electroporation, membrane thinning or thickening, inhibition of protein synthesis machinery, non-bilayer intermediates, modulation of anion carriers, inhibition of cell wall component biosynthesis, nucleic acid targeting, and non-lytic membrane depolarization [145, 146, 147, 148, 149, 150, 151]. The interactions of most cationic AMPs are with the bacterial cell surface. It occurs due to the negatively charged components embedded in lipid bilayers [152, 153]. Thus, the mechanism of action and activity of AMPs is influenced by amino acids chain length, overall charge of the peptide, structural conformation, amino acid composition, hydrophobicity, and amphipathicity [139]. These mechanisms are studied using different biophysical techniques like circular dichroism, FTIR spectroscopy, NMR, and other techniques and by performing various biological assays like cellular leakage assay, cell dyes, time-course of killing assay, and other assays [4, 15, 140, 141]. Lastly, the toxicity of the AMPs is determined to understand its safety for better treatments using different assays like the MTT assay, hemolytic assay, microscopic analysis, in vivo model study, serum stability, and other relevant toxicity assays [4, 15, 141, 154].
Therefore, to study the newly developed or designed AMPs against S. aureus and its resistant strains, the abovementioned experimental approaches, as illustrated in Fig. 4, must be performed with or without further amendments to understand the structure and biological activities of these AMPs and their possible biological applications.
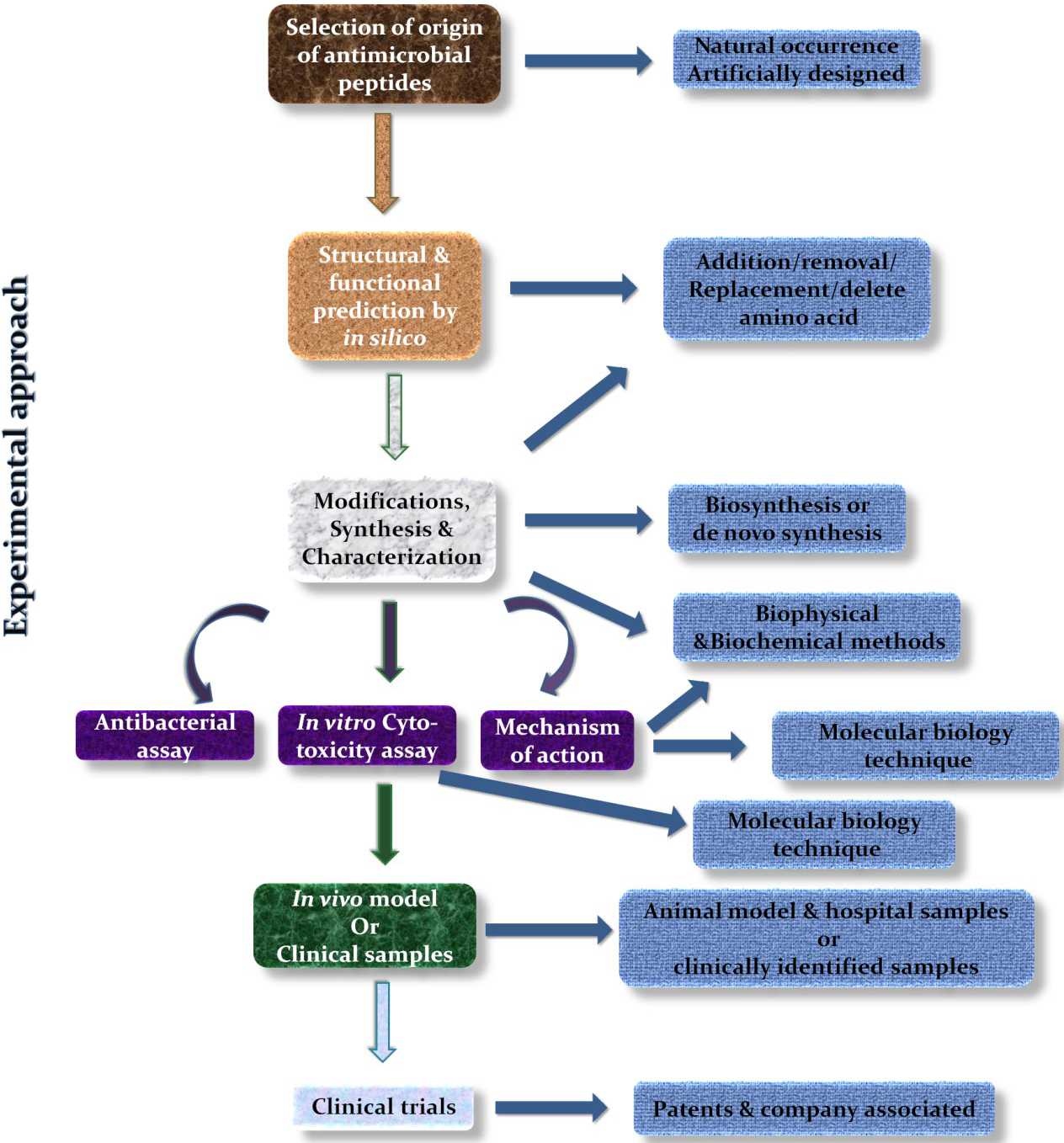 Fig. 4.
Fig. 4.Experimental strategy to study AMPs.
The infections caused by the S. aureus strain (i.e. not the resistant strains) can be treated by vancomycin, the last resort antibiotic against gram-positive bacteria. However, vancomycin can cause severe renal toxicity when used at extremely high concentrations. The toxicity also depends on the length of the treatments [157, 158]. Therefore, the infection by these resistant strains is becoming more difficult to treat. Moreover, the situation of community-acquired infections is steadily increasing. Thus, this situation had an impact on the clinical settings. Hence, new antibiotics or alternatives like AMPs are becoming highly necessary, however they must undergo clinical trials to explore their clinical activity. In our recent study, we designed the PBDM (Probiotic Bacteriocin Derived and Modified) peptides from a bacteriocin (m2163) present in Lactobacillus casei and synthesized it. Further, in silico, in vitro, and in vivo studies were performed with these peptides. These studies showed that the peptides were effective against MRSA, VRSA, and wild type S. aureusin vitro [15]. Thus, the shorter length forms of AMPs with modified sequences can be derived from other larger peptides displaying good antibacterial activity. Moreover, the list of peptides depicted in Table 1 shows the growing importance of the AMPs and its variations against S. aureus strains in 2020.
| Antimicrobial peptides | Origin | S. aureus and its variants | MIC value | Ref. |
| Enterocin EF35 | Enterococcus faecalis ECF35 | Staphylococcus aureus | [164] | |
| Enterocin TJUQ1 | Enterococcus faecium TJUQ1 | S. aureus | 46.50 |
[165] |
| CMP-Van-Lipo (Collagen mimetic peptide tethered vancomycin liposomes) | Synthetic | Methicillin Resistant S. aureus | van concentration (4 to 10 µg/mL) | [166] |
| HF-18 | Hagfish intestinal peptide | Drug resistant S. aureus | 4 µg/mL | [167] |
| HSER-CQDs | Retinoic acid receptor responder protein 2 and Carbon quantum dots | Vancomycin Resistant S. aureus | 25 µg/mL | [4] |
| Fr.A2 and Fr.B1 | Camel milk and cow milk | S. aureus | 130000 µg/mL | [168] |
| Backbone cyclized KR-12 dimers | Mammalian defense peptide LL-37 | S. aureus | 1.25 µM | [169] |
| Short cationic dialkyl lipopeptides (C10)2-KKKK-NH2and (C12)2-KKKK-NH2 | Synthetic | Methicillin susceptible S. aureus (MSSA), MRSA | 4000-16000µg/mL and 8000-32000 µg/mL | [170] |
| TC26 | Cyprinus carpio (C-terminal of tissue factor pathway inhibitor 1) | S. aureus | 10 µM | [171] |
| Alafosfalin; | Phosphonopeptides | S. aureus, MRSA and MSSA | 8 µg/mL; | [172] |
| di-alanyl fosfalin; | 16 µg/mL; | |||
| β-chloro-L-alanyl-β-chloro-L-alanine; | 2-4 µg/mL; | |||
| Fosfomycin | 16 µg/mL | |||
| Melittin (MEL); | Bee venom; | S. aureus | 5 µM; | [173] |
| MEL in presence of pyrrolidinium- based ionic liquids | Synthetic | 2-200 µM | ||
| Fermentaion of Kenaf seed protein produces bioactive peptides | Kenaf seed protein | S. aureus | 4000 µg/mL | [174] |
| Myticusin-beta | Mytilus coruscus | S. aureus | NA | [175] |
| AMP-CBP-FAM | Synthetic | S. aureus | [176] | |
| Melittin; | Apis mellifera | MRSA, entertoxin producer S. aureus SEC and SED | 7.2 µg/mL, 0.7 µg/mL and 3.6 µg/mL; | [177] |
| Apitoxin | 6.7 µg/mL, 7.2 µg/mL and 5.4 µg/mL | |||
| S7 (the most effective peptide) | Triplet-tryptophan-pivot peptides; Synthetic | S. aureus and MRSA | 4 and 2 µM | [178] |
| Bacteriocins (Bac22 and Bac4463) capped silver nanoparticle | Lactobacillus strains; Synthetic | S. aureus | 2 and 8 µg/mL | [179] |
| Teixobactin | Eleftheria terrae | Susceptible S. aureus and MRSA | 1 µg/mL | [180] |
| Indole-triazole-peptide conjugate (compound 9 series) | Synthetic | S. aureus | 10-16 µg/mL | [181] |
| Acetylated and non-acetylated QAK | Antheraea mylitta; Synthetic | S. aureus | 7.5 and 60 µg/mL | [182] |
| Intestinalin | Clostridium intestinale (LysC); Synthetic | S. aureus | 6 µg/mL | [183] |
| GAM019 (G19) and GIBIM-P5S9K (G17) peptide encapsulated on poly-lactic-glycolic-acid (PLGA) | Synthetic | MRSA | 0.7 and 0.2 µM | [184] |
| Analog of BSI-9 (compound 9-13;15) | BSI-9; Synthetic | S. aureus | 1-8 µg/mL | [185] |
| TC19 | Human thrombocidin-1-derived peptide | S. aureus | Lethal concentration (3.75 µM in PT buffer; 7.5-15 µM in PBS buffer; 15-30 µM in 50% plasma) | [186] |
| A1-A6; B1-B6 | Synthetic and modified version of A1 and B1 | MRSA | 1-8 µg/mL; 0.5-8 µg/mL | [187] |
| Melittin | Purchased from Mimotopes Peptide company (Mulgrave, Australia) | MRSA and MSSA | 0.5-8 µg/mL; 0.5-2 µg/mL | [188, 189] |
| Short self assembling cationic antimicrobial peptide mimetic library based on 3,5 diaminobenzoic acid scaffold (C2, C4, C6 and C7) | Synthetic | S. aureus | 27, 15.6, 29, and 3.9 µg/mL | [190] |
| RCP (Polypeptide enrich extract) | Skin of Rana chensinensis | S. aureus | 157.8 µg/mL | [191] |
| ID13; DLP4 | DLP4 (an insect defensin) | S. aureus | 4 µg/mL; 16 µg/mL | [192] |
| Pep-cur complex | Octaarginine (P) and cumin (C) | S. aureus | C= 3.0 ± 0.2 nmol | [193] |
| P = 5.2 ± 0.1 nmol | ||||
| PBDM1 and PBDM2 | Lactobacillus casei ATCC 334 (bacteriocin m2163) | S. aureus, MRSA and VRSA | 10-15 µg/mL; 10 µg/mL | [15] |
| OVTp12 | Ovotransferrin hydrolysate (egg) | S. aureus | 32 µg/mL | [194] |
| C |
Synthetic | S. aureus and MRSA | 3.90-31.25 µg/mL | [195] |
| Compound 14 | Amphiphilic sofalcone derivatives 1-17; Synthetic | S. aureus | 1.56 µg/mL | [196] |
| Adepamycin | Adenanthera pavonina seeds (sequence of plant trypsin inhibitor) | S. aureus | 1.8 µM | [197] |
| caP4 | Curcuma pseudomontana L. (Zingiberaceae) | S. aureus | 8 µg/mL | [198] |
| Bac8c; | Bovine (improved from Bac2A) | S. aureus and MRSA | 2 and 8 µg/mL; | [199] |
| Lipoic acid (LA) Bac8c | 1 and 4 µg/mL | |||
| Polydopamine/hydroxyapatite/nisin (PDA/HAP/Nisin) | Synthetic | S. aureus | 30 µg/mL | [200] |
| Natural peptide; S3K; G2K-S3K | Didymocentrus krausi (MK049518) | S. aureus | 12.5 µg/mL; | [154] |
| 3.13-6.25 µg/mL; | ||||
| 3.13-6.25 µg/mL | ||||
| P6.2 | Synthetic | S. aureus | 12.72 µM | [201] |
| ent A- col E1 | Enterocin A and Colicin E1; Synthetic | S. aureus | 10 µg/mL | [202] |
The innate immune system associated AMPs are good candidates for the development of the alternatives to antibiotics. However, they exert some cytotoxicity towards normal host cells that limits their inclusion as therapeutics. Thus, to overcome this hurdle, increased AMPs efficacy, stability, and reduced toxicity, strategies like structural analogs or modifications as well as combinations with conventional antibiotics or nanoparticles can be used as shown in Fig. 5 [155]. Zharkova et al., reported in their study that antibiotics with intercellular targets (e.g., rifampicin and gentamicin) and AMPs which are highly membrane-active (e.g., hBD-3 and protegrin 1) showed synergistic effects due to increased bioavailability [159]. Jelinkova et al., showed that the combination of Hecate peptide with vancomycin improved the efficacy of the components after forming the conjugate and reduced the overall toxicity. It allowed the treatment of VRSA, MRSA, and the non-resistant S. aureus [141]. In another study, we used the combination strategy by combining the HSER peptide (derived from retinoic acid receptor responder protein 2) with carbon quantum dots (CQDs) and tested this combination against resistant strains of S. aureus and E. coli. The results revealed that the HSER-CQDs were more effective at a lower concentration in comparison to the individual components. The HSER-CQDs was found to be non-toxic and hemocompatible [4]. Therefore, these combinatorial strategies help to overcome resistance, reduce dosage, enhance the selectivity of the compound, and reduce side effects [160, 161, 162]. AMPs can also be used for targeted delivery or can be encapsulated within some nano-capsule to reduce their toxicity, proper release, and increase stability [156]. Thus, AMPs are molecules which can be modified in various ways to improve their activity for therapeutic purposes based on clinical demands.
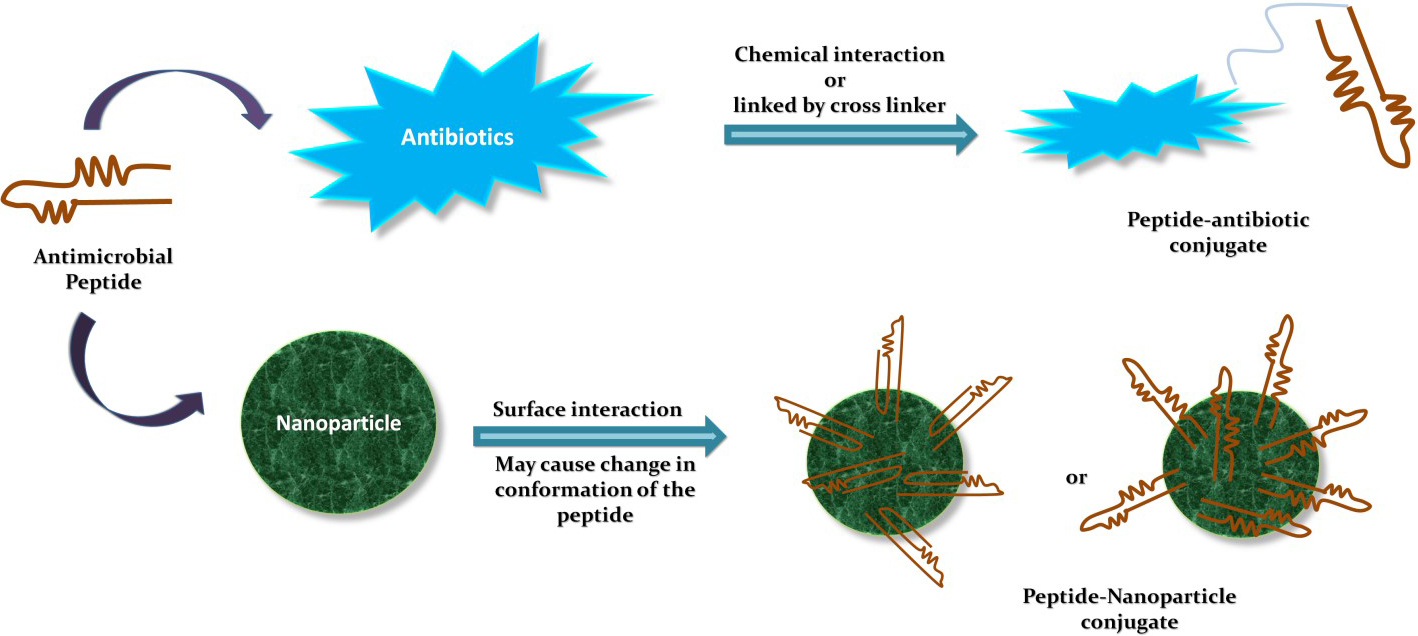 Fig. 5.
Fig. 5.Combinatorial strategies using AMPs.
Moreover, the studies on in vivo infection models helped to understand the efficacy of the AMPs for treating infections in the mammalian systems. At the beginning of this section, we discuss PBDM peptides. An in vivo study with VRSA infected BALB/c mice model showed that these PBDM peptides were able to treat VRSA infection and cure them without visible side effects [15]. Another study by Fan Yu, et al., showed a new concept of entrapping bacteria instead of killing by using an HDMP (human defensin-6-mimic peptide). HDMP inhibits bacterial invasion by trapping bacteria using its self-assembled fibrous network. This mechanism was apparent with both S. aureus- and MRSA-infected BALB/c mouse model. However, HDMP is active only against gram-positive bacteria but not against gram-negative bacteria [163]. Thus, these in vivo studies helped to understand the activity of this peptide in the live system. These in vivo studies will help to determine the future of these peptides whether it will be heading towards the clinical studies or towards the need to generate modified peptides or to use these peptides as conjugates or entrap them in a capsule to reduce their toxicity and targeted delivery.
Although a great deal of research is focused on AMPs, only a very small number of AMPs is in clinical use when compared to conventional antibiotics. Some of the peptides that are undergoing clinical trials are Pexiganan (MRSA,) LTX-109 (against nasal colonies of MRSA/MSSA), MU1140 (MRSA), NP432 (MRSA), and AP138 (MRSA implant infections). Most of them are undergoing preclinical evaluation excluding Pexiganan which is undergoing Phase III clinical testing whereas LTX-109 is under clinical phase I/II. Even if the whole process of clinical trials takes more than 8 years (Phase I 1.5 years; Phase II 2 years; Phase III 3 years; introduction to the market 1.5 years; Phase IIIb/IV- for further clinical use and re-confirmations of its use) to eventually bring a drug into the market for proper medical use [203]. Therefore, it is necessary to launch a greater number of clinical trials to bring out the potential of AMPs in the world of therapeutics against the ever growing bacterial infections especially with antibiotic resistant bacteria.
The infections caused by the MRSA and VRSA strains have become a serious health issue due to their resistance to antibiotics. Thus, the treatments against these strains are prolonged and/or can end up in a failure. The discussion about their mechanisms of antibiotic resistance will not only help the researchers to target specific genes and proteins for the development of new therapeutics but it will also help them to further investigate the unsolved tasks for a better understanding of the pathogenesis.
Though many efforts are aimed at producing new antibiotics, the choice of an alternative to antibiotics can also be an interesting approach. There are reports of resistance towards AMPs which can lead to the development of future resistance to host AMPs. However, this can be overcome by synthetically modified peptides [156, 204]. Apart from this, the limitation of its high cost of production, degradation by proteases, toxicity, and their unknown pharmacokinetics can also be a problem [205]. However, these can be solved by the use of relatively shorter peptides that will reduce the overall production costs. The use of different drug delivery systems can reduce their toxicity, maintain proper release, and improve stability. They can also be used in combination with antibiotics for better activity and can overcome resistance. Thus, combination strategies for therapy will become the modality of choice to overcome antibiotic resistance. Despite their limitation, the growing interest in AMPs as an alternative to antibiotics against S. aureus will improve its status in the future for developing new therapeutics. It will be an interesting subject of research to explore these AMPs as an alternative to antibiotics not only against the S. aureus but also against other pathogens.
M. conceived and designed the framework of the articles, wrote the paper, prepared the figures and thoroughly checked the paper; V.A. supervised the work and checked the paper for detail correction.
Thanks to all the peer reviewers and editors for their opinions and suggestions.
This work was financially supported by ERDF “Multidisciplinary research to increase application potential of nanomaterials in agricultural practice” (No. CZ.02.1.01/0.0/0.0/16_025/0007314) and CEITEC 2020 (LQ1601) with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustainability Programme II is gratefully acknowledged.
The authors declare no conflict of interest.