
Pulmonary arterial hypertension (PAH) is a progressive and fatal lung disease of multifactorial etiology. Most of the available drugs and FDA-approved therapies for treating pulmonary hypertension attempt to overcome the imbalance between vasoactive and vasodilator mediators, and restore the endothelial cell function. Traditional medications for treating PAH include the prostacyclin analogs and receptor agonists, phosphodiesterase 5 inhibitors, endothelin-receptor antagonists, and cGMP activators. While the current FDA-approved drugs showed improvements in quality of life and hemodynamic parameters, they have shown only very limited beneficial effects on survival and disease progression. None of them offers a cure against PAH, and the median survival rate remains less than three years from diagnosis. Extensive research efforts have led to the emergence of innovative therapeutic approaches in the area of PAH. In this review, we provide an overview of the current FDA-approved therapies in PAH and discuss the associated clinical trials and reported-side effects. As recent studies have led to the emergence of innovative therapeutic approaches in the area of PAH, we also focus on the latest promising therapies in preclinical studies such as stem cell-based therapies, gene transfer, and epigenetic therapies.
Pulmonary arterial hypertension (PAH) is a progressive and fatal lung disease of multifactorial etiology (Montani et al., 2013) (Fig. 1). The 2015 pulmonary hypertension guidelines of the European Society of Cardiology (ESC)/European Respiratory Society (ERS) and the 6th WSPH congress Task Force (Nice, 2018) revised the definition of PAH (Condon et al., 2019; Galie et al., 2016, 2019). Consequently, PAH is now hemodynamically defined as a mean pulmonary arterial pressure (mPAP) ≥ 20 mmHg at rest as assessed by right heart catheterization (RHC) and a pulmonary vascular resistance ≥ 3 Wood Units in the definition of all forms of pre-capillary pulmonary hypertension (PH) (Condon et al., 2019; Galie et al., 2016, 2019). Recent data showed that the normal mPAP is 14 ± 3 mmHg at rest with an upper limit of normal of approximately 20 mmHg. The clinical significance of a mPAP between 21 and 24 mmHg remains unclear. In PAH, increased pressure in the vessels is associated with pulmonary artery vascular remodeling, obstruction of small arteries (Lai et al., 2014), and an increase in the vascular resistance to blood flow through the lungs. Over time, high blood pressure can damage the heart and lead to RV failure and, ultimately, death (Fig. 2). PAH is a rare condition, which affects about 15-50 people per million in the United States.
 Figure 1.
Figure 1.Updated classification of pulmonary hypertension. This classification is based on recommendations from the 5th World Symposium on Pulmonary Hypertension in Nice, France, 2013. The World Health Organization classifies PAH into five broad groups of pulmonary hypertension based on the clinical similarities. The five groups of PH are: 1-PAH, 2-PH associated with left heart disease, 3- PH associated with chronic lung disease and/or hypoxia, 4- chronic thromboembolic PH (CTEPH) and 5- PH with unclear or multifactorial mechanisms.
 Figure 2.
Figure 2.Pathogenesis of pulmonary hypertension. PAH is characterized by a vascular remodeling of distal pulmonary arteries, vasoconstriction, endothelial dysfunction, inflammation and thrombosis leading to the formation of plexiform lesions. Proliferation and migration of pulmonary endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs) contribute to the vascular muscularization and obstruction, which progressively increase vascular resistance in PAH and induce right ventricle (RV) hypertrophy and ultimately RV dysfunction.
Pathophysiologic characteristics of PH include vasoconstriction, thrombosis and inflammation. Pulmonary vascular remodeling is associated with smooth muscle and endothelial cells dysfunction (Bisserier et al., 2020; Shimoda and Laurie, 2013). It is now well established that pulmonary artery endothelial cells (PAECs) play a major role in the regulation of vascular tone, inflammation, and thrombosis. Indeed, abnormal endothelial cell proliferation, along with exuberant neoangiogenesis, leads to the formation of glomeruloid structures known as plexiform lesions. They are described as common pathological features of the pulmonary vessels of patients with PAH. In addition, the proliferation and migration of the pulmonary artery smooth muscle cells (PASMCs) contribute to the muscularization of the small pulmonary arteries and, therefore, to vascular obstruction, which progressively increases vascular resistance in PH (Shimoda and Laurie, 2013).
The National Institutes of Health (NIH) registry on primary PAH reported that the median survival was 2.8 years between 1980-1991 when no specific and efficient treatments were available (D'Alonzo et al., 1991). Recently, the Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL) investigators showed a significant increase in survival of PAH patients in an era (2006-2012) with the current treatments (McGoon and Miller, 2012). The median survival now approximates 7 years in the modern treatment era. Despite the available treatments and ongoing research efforts, there is currently no curative treatment curative treatment against PAH.
Signs and symptoms of pulmonary hypertension are often subtle and nonspecific, and therefore not usually detected in a routine physical examination. Clinical suspicion of PH is based on symptoms, evaluation of risk factors, and a careful examination of the patient's medical and family history is critical for the diagnosis of PH. The most common symptoms include exertional dyspnea, fatigue, weakness, light-headedness/syncope, and chest pain. At more advanced disease stages, signs and symptoms of cardiovascular abnormalities are often detected and associated with heart and lung conditions. Several established diagnostic and screening tools are being used to confirm the presence of PH. The electrocardiogram (ECG) is a non-invasive and reliable test that may be used to reveal abnormalities of the electrical cycle within the heart of patients with PH. An ECG may demonstrate the presence of RV enlargement, such as tall right precordial R waves, right axis deviation, and right ventricular strain. Confirmation of pulmonary hypertension is based on the presence of tricuspid regurgitation. As a normal ECG does not exclude PH, additional tests are necessary to define the diagnosis.
The two-dimensional echocardiography with Doppler flow remains the most critical non-invasive screening tool to establish the diagnosis. An echocardiogram exam is of great importance in the diagnosis of PH as it will help to evaluate the RV enlargement and thickness, the tricuspid regurgitant velocity, the pulmonary vascular resistance as well as pressure and diameter of the pulmonary artery. If PH is suspected, further examination will help to define the underlying etiology and, therefore, the best treatment. Among them, genetic tests, pulmonary function tests and assessment of arterial blood oxygenation, high-resolution computerized tomography, chest X-ray, cardiopulmonary exercise testing (CPET), and cardiac catheterization are essential to determine the underlying cause of PH.
A right heart catheterization (RHC) remains the gold standard in diagnosing and defining the foundation of PH. In fact, RHC is the only diagnostic technique for reliably and accurately confirming the existence of PH as it provides a direct and accurate measurement of the RV and pulmonary artery pressure. During this procedure, a catheter is threaded in the RV through a central vein. More recently, the World Symposium on Pulmonary Hypertension suggested that the newly hemodynamic threshold should include a mean pulmonary artery pressure > 20 mmHg, a pulmonary artery wedge pressure < 15 mmHg, and a pulmonary vascular resistance of > 3 Wood units for pre-capillary PH. The complementarity of these afore-mentioned tests allows physicians to determine the underlying etiologies of PH and identify the best therapeutic options (Barst et al., 2004).
Nowadays, ongoing research aims to identify new non-invasive screening tool for the early diagnosis of PH and include the search of new biomarkers. The identification of potential early PH biomarkers may offer new opportunities for early therapeutic interventions and improve the quality of life as well as increase life expectancy.
Indeed, PH is classified into five groups by the World Health Organization (WHO) based on the underlying pathological and hemodynamic characterization (Simonneau et al., 2013) (Fig. 1). According to the WHO classification, Group I (PAH) refers to idiopathic PAH, heritable associated with genetic alteration such as BMPR2 (most common mutations in PAH), ALK1, Endoglin, SMAD9, CAV1, KCNK3 (less common mutations), drug and toxin-induced (amphetamines, methamphetamines, cocaine, fenfluramine-phentermine), and PH-associated with other systemic diseases (connective tissue diseases, HIV infection, portal hypertension, congenital heart disease) (Simonneau et al., 2013). PH due to left heart disease (left ventricular systolic dysfunction, left ventricular diastolic dysfunction, valvular heart disease, left heart inflow and outflow obstructions) are included in the Group II (Simonneau et al., 2013). PH in group III is due to lung diseases such as chronic obstructive pulmonary disease (COPD), interstitial lung diseases such as pulmonary fibrosis, sleep-disorders, alveolar hypoventilation disorders, chronic high altitude exposure and developmental abnormalities of the lung (Simonneau et al., 2013). Group IV refers to chronic thromboembolic pulmonary hypertension (CTEPH), which may occur when blood clots affect the flow of blood in the pulmonary arteries and other chronic pulmonary artery obstructions (angiosarcoma, arteritis, etc...) (Simonneau et al., 2013). Finally, all forms of PH caused by unclear, unknown, or multifactorial mechanisms are included in Group V (Simonneau et al., 2013). This large and diverse group includes PH caused by hematologic disorders, systemic disorders, metabolic disorders, chronic renal failure, tumor-associated, and other rare diseases.
Although many molecular pathways have been identified over the past decades in the pathogenesis of PH, all currently approved therapies target mostly three significant pathways: prostacyclin, endothelin (ET), nitric oxide (NO) (Sitbon and Morrell, 2012). Indeed, most of the available and FDA-approved treatments for treating PH are exclusively recommended to WHO Group I PH patients and fall into four categories: a) Prostacyclin Analogues and receptor agonists, b) Phosphodiesterase 5 inhibitors, c) ET receptor antagonists, d) cGMP activators (Stamm et al., 2011) (Fig. 3). The only FDA-approved therapy for use in chronic thromboembolic PH (CTEPH) is riociguat (Ghofrani et al., 2010). In this review, we provide an overview of the current FDA-approved therapies in PH and discuss the associated-side effects. As intensive research efforts have led to the emergence of innovative therapeutic approaches in the area of PH, we also focus on the latest emerging and promising therapies in preclinical studies such as stem cell-based therapies, gene therapies, and epigenetic therapies.
 Figure 3.
Figure 3.Pathways targeted by the current FDA-approved PAH therapies. FDA-approved PAH therapies target mostly three major pathways: prostacyclin, endothelin (ET), nitric oxide (NO). They fall into four categories: 1- Prostacyclin Analogues and receptor agonists (epoprostenol, tetroprostinil, iloprost, selexipag), 2- Phosphodiesterase 5 inhibitors (sildenafil, tadalafil), 3- ET receptor antagonists (ambrisentan, bosentan, macitentan), 4- cGMP activators (riociguat).
Prostacyclin (also called prostaglandin I2 or PGI2) is a potent vasodilator and inhibitor of platelet aggregation. Prostacyclin is produced from prostaglandin H2, which is catalyzed by the enzyme prostacyclin synthase in endothelial cells. It binds to G protein-coupled receptors located on PASMCs and activates adenylates cyclase, inducing vasodilatation of PASMCs via inhibition of myosin light chain kinase (MLCK) (Majed and Khalil, 2012). In addition to its vasodilator properties, prostacyclin also inhibits proliferation and protects against pulmonary remodeling. Given these anti-proliferative and vasodilators properties, the role of prostacyclin in PAH has been investigated (Fig. 3). Several prostacyclin analogs were developed for the treatment of PAH and have since revolutionized the treatment of this disease. There are currently three approved prostacyclin analogs for the treatment of WHO Group 1 PH (including idiopathic PAH, hereditary PAH, and connective tissue related disease): epoprostenol, treprostinil, and iloprost (Gomberg-Maitland and Olschewski, 2008).
4.1.1 Epoprostenol
Epoprostenol is a synthetic prostacyclin (PGI-2) analog. Epoprostenol was the first approved drug for the treatment of PAH in 1995 and remains the only medication showing a significant decrease in mortality in iPAH as of today (Sitbon and Vonk Noordegraaf, 2017). Substantial evidence, from both randomized control trials and observational studies, is available to support the use of epoprostenol. Two crucial randomized controlled trials investigated the consequences of epoprostenol on hemodynamic and clinical outcomes in patients with PAH (Barst et al., 1996; Rubin et al., 1990). The first was performed by Rubin et al., evaluating the effects of epoprostenol versus standard care (included digoxin, diuretics, and warfarin and calcium channel blockers at the time) in 23 patients with "primary pulmonary hypertension". They found that administration of epoprostenol, via continuous intravenous infusion, significantly reduced pulmonary vascular resistance at 2 months, with a decrease > 10mmHg of the mean pulmonary artery pressure in six out of ten patients in the treatment group, compared with one of nine in the control-group (despite the absence of statistical significance) (Rubin et al., 1990).
In 1996, Barst and colleagues conducted a similar randomized control trial to evaluate the effects of epoprostenol at 12 weeks in 81 patients with NYHA stage III-IV Primary PH (Barst et al., 1996). The investigators found significant improvements in exercise capacity, quality of life, and hemodynamic variables such as mPAP, cardiac index, and PVR in the epoprostenol group at 12 weeks. Importantly, it also showed an improvement in survival in the group that received Epoprostenol (Barst et al., 1996).
Epoprostenol has also been studied in Scleroderma related PH. In a randomized control trial conducted by Badesch et al., 111 patients with moderate to severe PH related to scleroderma spectrum of disease were randomized to receive epoprostenol infusion in combination with conventional therapies versus conventional therapies alone (Badesch et al., 2000). Their results showed an improvement in the exercise capacity in patients receiving epoprostenol. In contrast, it declined in those receiving only the traditional treatments at 12 weeks (difference of 108m in the median distance walked). Improvement in hemodynamics (mean PAP and PVR) and Borg dyspnea score were reported in the group receiving epoprostenol (Badesch et al., 2000). Following the results of the afore-mentioned trial, epoprostenol has been approved for patients with WHO Group 1 PAH with NYHA functional class III-IV symptoms. Pharmacokinetic properties of epoprostenol affect the method of administration. Indeed, epoprostenol is a highly temperature-sensitive drug with a short half-life of only 6 minutes at 37°C. Therefore, its delivery occurs via continuous intravenous infusion (Barst, 2010). It causes significant flushing in patients, which is very uncomfortable for the patients. This delivery method requires permanent intravenous access, patient competence in self-administration, and a reliable home support system. Sudden discontinuation is associated with further clinical implications, as prompt restoration of epoprostenol infusion is necessary to prevent the development of pulmonary hypertension and circulatory collapse. A high degree of patient adherence to therapy is needed.
A recent formulation of epoprostenol (epoprostenol AM) with mannitol and arginine excipients showed a longer half-life at room temperature and has allowed for greater ease of administration (Lambert and Bandilla, 2012). Unfortunately, commonly reported side effects related to the medication include jaw pain, headache, nausea, vomiting, and flushing. Intravenous delivery system-related adverse events were also reported, such as catheter-related infection and thrombosis (Badesch et al., 2009).
4.1.2 Treprostinil
Treprostinil was the next prostacyclin analog to be approved by the FDA for its use in PAH. Its stability at neutral pH, room temperature, and extended half-life of 4.5 hours allowed for more flexibility regarding the delivery methods and storage. Treprostinil can also be subcutaneously administrated, which decrease catheter-related complications such as thrombosis and infection (Simonneau et al., 2002; Ventetuolo and Klinger, 2012). The randomized control trial, leading to its FDA-approval in 2002, included 470 patients with NYHA II-IV PAH (including primary pulmonary HTN, Connective tissue disease-related PH, or associated with congenital heart disease). The study randomized patients to receive either a subcutaneous treprostinil infusion in combination with conventional therapy or conventional therapy in combination with a placebo for 12 weeks. Interestingly, treprostinil was associated with a modest increase of 16m in median 6MWD, along with significant improvements in symptoms, quality of life, and cardiac hemodynamics, including mean PAP, PVR, and cardiac index (Simonneau et al., 2002). The most commonly observed side effect was infusion-site pain (85% in treprostinil vs. 27% in control), which is also a limiting factor for its use in clinical practice. Jaw pain, diarrhea, flushing, and pedal edema were also reported.
In 2006, an uncontrolled multicenter study evaluated the efficacy and safety of intravenous infusion of treprostinil in a similar PAH population. Tapson and colleagues found that treprostinil improved the mean 6MWD by 82m, Borg Dyspnea scale scores, and cardiac hemodynamics, without any serious adverse effects (Tapson et al., 2006). Besides, the TRIUMPH trial demonstrated that treprostinil could be delivered via inhalation as add-on therapy in patients with group 1 PAH McLaughlin et al. (2010). This study randomized 235 patients NYHA III-IV with PAH, who were already on bosentan or sildenafil, to receive four inhalations of treprostinil per day vs. placebo. An improvement in the median 6MWD by 20m (tested 4hours after inhaler use) at 12 weeks was reported in the treatment group, with a significant increase in the quality of life scores McLaughlin et al. (2010). Inhaled vasodilator therapy showed several benefits, such as limited side effects given local delivery and decreased the overall systemic dose. However, the use of treprostinil as monotherapy is yet to be investigated.
4.1.3 Iloprost
Iloprost was developed as an inhaled prostacyclin analog. The "aerosolized iloprost randomized" study, known as AIR, was a randomized control trial that evaluated the safety and efficacy of inhaled iloprost as monotherapy in 203 patients with severe PAH and inoperable Chronic thromboembolic pulmonary hypertension- NYHA functional class III-IV over 12 weeks (Olschewski et al., 2002). The co-primary endpoint of improvement of at least one NYHA class and 10% improvement in 6MWD was met by 16.8% of patients receiving iloprost compared with 4.9% patients receiving a placebo. The major change in the six-minute walk test was also greater in the group who received iloprost when compared to the placebo, as was dyspnea index and quality of life scores. Hemodynamics, including PAP, PVR, and cardiac index, were all largely unchanged from the baseline before the first iloprost administration, while significant worsening was noted in the placebo group (Olschewski et al., 2002). Side effects were mild and included flushing and jaw pain. Although syncope was reported in both groups, they were often categorized as more severe in the Iloprost group.
In 2006, the STEP study evaluated the efficacy of inhaled iloprost as an add-on to bosentan monotherapy in 67 patients with PAH (McLaughlin et al., 2006). This study revealed significant improvements in 6MWD, NYHA functional class, and delayed time to clinical worsening compared to bosentan monotherapy alone (McLaughlin et al., 2006). In conclusion, the previous studies have confirmed that iloprost is safe and efficient as an add-on therapy. Frequent dosing makes practical use challenging for patients.
4.1.4 Selexipag
Selexipag is another oral selective-agonist of prostacyclin receptors. The GRIPHON trial was a phase 3 randomized control trial in which 1156 patients with WHO group I PAH were randomized to receive selexipag or placebo (Sitbon et al., 2015). The enrolled patients were initially either without any treatment or receiving an Endothelin Receptor Agonist, PDE-5 inhibitor, or both for at least 90 days. The risk of primary composite endpoint of death and disease progression was significantly decreased in patients receiving selexipag. No difference in mortality was reported between the two groups. Frequent adverse events leading to discontinuation, including headache, diarrhea, and nausea, were noted (Sitbon et al., 2015). Oral administration circumvents the complications associated with permanent intravenous access.
NO diffuses into the cytoplasm of underlying smooth muscle cells and activates soluble guanylate cyclase (sGC), increases the production of the second messenger cyclic guanosine monophosphate level (cGMP) and subsequently activates cGMP-dependent protein kinase (PKG) (Forstermann and Sessa, 2012). Activation of PKG stimulates Ca2+-activated potassium channels, which leads to membrane hyperpolarization and inhibition of the regulatory subunits of myosin light chain phosphatase and phosphorylation of myosin-binding proteins. In the lung, cGMP is metabolized by the type 5 isoform of cyclic nucleotide phosphodiesterase (PDE-5) (Wilkins et al., 2008). PDE-5 is also expressed in vascular smooth muscle cells, myocardium, and platelets (Wilkins et al., 2008). As previously mentioned, cGMP is an essential regulator of the pulmonary vascular tone as a secondary messenger of nitric oxide. It also regulates SMC proliferation and apoptosis (Wharton et al., 2005). Upregulation of PDE-5 has been noted in the lungs of patients with PAH, leading to decreased levels of cGMP and increased intracellular Ca2+, which is associated with increased vasoconstriction and abnormal proliferation (Montani et al., 2009; Ventetuolo and Klinger, 2012). The inhibition of the PDE-5 activity has been extensively investigated in the treatment of PAH. Three PDE-5 inhibitors have been FDA-approved for its use in PAH: sildenafil, tadalafil, and vardenafil (Buckley et al., 2010) (Fig. 3).
4.2.1 Sildenafil
Sildenafil was the first PDE-5 inhibitor to be approved following the results of the SUPER randomized controlled trial, which evaluated the efficacy and safety of three doses of sildenafil (20mg TID, 40mg TID, and 80mg TID) in patients with PAH (idiopathic, connective tissue disease and congenital systemic to pulmonary shunts) (Rubin et al., 2011). This trial showed a significant improvement in exercise capacity, assessed by 6MWD for all three sildenafil doses vs. placebo. At 12 weeks, 6MWD was 45m, 46m, and 50m for 20mg, 40mg, and 80mg of sildenafil, respectively (P < 0.001). No significant inter-dose differences were noted (Rubin et al., 2011). A significant improvement in WHO functional class and hemodynamic measurements (mPAP, PVR, and CI) were also described in patients receiving sildenafil at 12 weeks. Headache, flushing, and diarrhea were among the reported side effects. Interestingly, 46% of the patients enrolled in the extension study with sildenafil 80mg TID showed a sustained improvement in the 6MWD, and 60% maintained or improved functional status (Galie et al., 2005; Rubin et al., 2011). Given the lack of significant improvements after the original 12-week trial, the FDA has only approved the 20mg dose for PAH.
4.2.2 Tadalafil
The efficacy and safety of tadalafil were investigated in the PHIRST trial, which was a randomized control trial comparing different doses of tadalafil (2.5mg, 10mg, 20mg, 40mg per day) versus placebo in patients with PAH (including idiopathic/heritable, anorexigenic use related, HIV infection, congenital to systemic shunt) (Galie et al., 2009; Oudiz et al., 2012). Of note, patients on any prior therapy (except bosentan at a stable dose of 125mg per day) were excluded from the study. A significant increase in the 6MWD was achieved with 40mg tadalafil at 33m. Considerable improvement in the quality of life scores and time to clinical worsening was observed with tadalafil 40mg a day, although > 50% of all patients were on bosentan. 93 patients underwent hemodynamic assessment, with improvement in mPAP and PVR when treated with 20mg and 40mg of tadalafil. Additionally, improvement in the cardiac index in the 40mg group was noted (Galie et al., 2009).
4.2.3 Vardenafil
Although vardenafil has so far been used and approved for the treatment of erectile dysfunction, it has been shown to be useful in PAH as well (Jing et al., 2011). A randomized control trial conducted in China investigated the use of vardenafil 5 mg twice daily in 66 PAH patients WHO functional class II-III (including idiopathic, connective tissue disease-related, congenital systemic to pulmonary shunts). Only patients who were not on any PH-specific therapies for 3 months before enrollment were included (Jing et al., 2011). A significant improvement in the median 6MWD of 69m was seen in patients receiving vardenafil, which was maintained during the extension phase at 24-weeks. Improvement in hemodynamics and symptoms were also noted. However, further trials confirmed that generalization and validation in other races remain necessary (Jing et al., 2011). This class of medications is generally well-tolerated. Some of the side effects-observed were headache, nausea, myalgias. Visual side effects have been noted with PDE5 inhibitors in the Erectile Dysfunction trials but not during the PAH trials (Buckley et al., 2010).
4.2.4 Riociguat
Riociguat is a soluble guanylate cyclase stimulator increasing the cGMP availability and also acts in synergy with nitric oxide. A phase 2 uncontrolled open-label clinical trial primarily evaluated the safety profile of riociguat in 42 patients with chronic thromboembolic pulmonary hypertension (CTEPH) and 33 patients with PAH. The investigators found a significant improvement in 6MWD and hemodynamics in both groups (though this was a secondary endpoint) (Ghofrani et al., 2010). Another phase 3 trial aimed to evaluate its safety and efficacy in 443 patients with PAH who were randomized to receive riociguat or placebo. At 12 weeks, they observed a significant improvement in placebo-adjusted mean 6MWD of 36 m along with a substantial improvement in hemodynamic variables (Ghofrani et al., 2013). The FDA subsequently approved riociguat for its use in patients with CTEPH and PAH.
ET-1 is a 21 amino-acid peptide predominantly secreted by the vascular endothelial cells. ET-1 is mostly described as a potent vasoconstrictor peptide that can constrict blood vessels and contribute to vascular remodeling (Hynynen and Khalil, 2006). ET-1 is released by exocytosis in response to various stimuli such as hypoxia, growth factor, mechanical stretch, cytokines, and adhesion molecules. Its effects are mediated through two types of receptors: endothelin A receptor (ETA) and the endothelin B receptor (ETB). Activation of ETA receptors, predominantly expressed in smooth muscle cells, leads to vasoconstriction and cell growth. Their activation potentiates pulmonary vascular remodeling (Hynynen and Khalil, 2006). ETB is only located in the vascular ECs, and their activation promotes the production of NO and PGI2, leading to vasodilation. The exclusive distribution of ETB on EC is associated with specific preclinical effects and remains of particular importance for the design of optimal pharmacological strategies to antagonize the ET system. Therefore, blocking the activity of ET-1 receptors, especially the ETA receptors, may be a promising therapeutic strategy. Three ET-receptor antagonists have been FDA-approved for clinical use: bosentan, ambrisentan, and macitentan (Fig.3). Bosentan and macitentan are dual ETA and ETB-receptor antagonists, whereas ambrisentan has higher selectivity toward the ETA receptors (Wei et al., 2016).
4.3.1 Bosentan
Bosentan was the first dual endothelin receptor antagonist to be approved by the FDA in 2001 for NYHA III-IV WHO group 1 PH after the results of the BREATHE-1 randomized control trials. This clinical trial randomized 213 patients with iPAH or Connective Disease-related PH to receive 62.5 mg bosentan for 4 weeks, followed by 125 mg or 250 mg bosentan daily for 12 weeks or placebo (Channick et al., 2001; Rubin et al., 2002). There was an average difference of 44m in the 6MWD between combined treatment groups and placebo group at 16 weeks, which was statistically significant along with an increase in time to clinical worsening, favoring bosentan use (Rubin et al., 2002).
4.3.2 Ambrisentan
Ambrisentan, a selective ETA-antagonist, was approved in 2013, after the ARIES-1 and ARIES-2 trials. These two concurrent randomized control trials used different doses of ambrisentan (Galie et al., 2008). Patients with severe PAH (including idiopathic, related to connective disease, HIV infection, or anorexigenic use) were randomized to receive a placebo or 5 mg or 10 mg (in ARIES-1) or 2.5 mg or 5 mg (in ARIES-2) of ambrisentan. The placebo-corrected 6MWD was significantly higher in the group receiving ambrisentan in both ARIES-1 and ARIES-2: 31m for 5 mg and 51m for 10 mg of ambrisentan for ARIES-1 and 32m for 2.5 mg and 59m for 5 mg of ambrisentan for ARIES-2. A statistically significant decrease in time to clinical worsening was noted in ARIES-2 in patients receiving ambrisentan compared to placebo. Improvement in functional class was also indicated in ARIES-1 (Galie et al., 2008). Ambrisentan has the advantage of a longer half-life, allowing for once-daily dosing (compared to bosentan).
4.3.3 Macitentan
Macitentan is a dual ET-receptor antagonist and was developed by modifying the structure of bosentan to improve efficacy and safety. It was FDA approved in 2013 based on the results obtained from the SERAPHIN trial. It was one of the longest trials at the time to assess the safety and efficacy of an endothelin receptor antagonist with an evaluation of morbidity and mortality as primary end-points (Pulido et al., 2013). 742 patients with WHO functional class II-IV PAH (including idiopathic PAH, heritable PAH, connective tissue disease-related, HIV infection, secondary to repaired systemic to pulmonary shunts, drug/toxin-induced) were randomized to receive placebo, macitentan 3 mg or macitentan 10 mg (Pulido et al., 2013). At 115 weeks, 46.4% of the placebo group, 38% of 3 mg macitentan group and 31.4% of the 10 mg macitentan group have achieved the composite primary endpoint of time from initiation of treatment to first event-related to PAH (including worsening of PAH, initiation of prostanoid therapy, lung transplantation or atrial septostomy) (Pulido et al., 2013). The hazard ratio for the primary endpoint was 0.70 with 3 mg macitentan vs. placebo and 0.55 for 10 mg macitentan vs. placebo (Pulido et al., 2013). Improvement in the 6MWD and hemodynamic parameters was also noted in the group receiving macitentan vs. placebo. The daily dosing regimen of this drug is also advantageous.
The primary safety concern with ERA's is a transient and reversible transaminitis, first reported in the BREATHE-1 trial with bosentan (Denton et al., 2008). This initially led to mandated monthly hepatic function monitoring for patients under treatment. However, the SERAPHIN trial, which was the longest monitoring trial, did not identify similar side effects on the liver. Also, the incidence of transaminase elevation was similar in placebo and ERA groups in the ARIES-1/2 trials, following which the FDA lifted this mandate (Oudiz et al., 2009; Ventetuolo and Klinger, 2012). A higher percentage of anemia was also reported in patients receiving macitentan. Sitaxsentan, another ETA-selective antagonist, showed initial promising results given its receptor-specificity. Unfortunately, this new drug was withdrawn from the market after several cases of fatal idiosyncratic hepatotoxicity (Chin et al., 2012).
Levosimendan functions as a calcium sensitizer by increasing the affinity of myocardial troponin C to calcium, without increasing the level of intracellular calcium, and thus exhibits positive inotropic properties in myocytes (Kass and Solaro, 2006). In addition, the cardioprotective effects of levosimendan are also mediated through its action on the opening of the mitochondrial (ATP)-sensitive potassium channels (Kass and Solaro, 2006). Interestingly, levosimendan promotes vasorelaxation by facilitating the opening of adenosine triphosphate (ATP)-sensitive potassium channels in smooth muscle cells (Kass and Solaro, 2006). Given the inotropic properties of levosimendan, its clinical use is indicated for the management of acutely-decompensated severe congestive heart failure, when no conventional therapy is available to the patient (Nieminen et al., 2013). The HELP (Hemodynamic Evaluation of levosimendan in Patients with PH-HFpEF) trial is a 6-week double-blind placebo-controlled phase 2 trial, which aims to evaluate the effectiveness and safety of intermittent levosimendan compared with placebo in patients with pulmonary hypertension and heart failure with preserved ejection fraction (PH-HFpEF). Recent preliminary data showed a significant improvement in pulmonary capillary wedge pressure (PCWP) during supine exercise, with a mean reduction of 8 mmHg in the patients treated with levosimendan. PCWP is an indirect measure of the left atrial pressure and is considered the gold standard for determining the cause of acute pulmonary edema. Furthermore, most patients showed a significant decrease in pulmonary arterial pressure of an average of 4.9 mmHg.
Simonneau et al. have investigated in a randomized clinical trial, called PACES-1, the therapeutic potential of combination therapy (Simonneau et al., 2008). As oral sildenafil and intravenous epoprostenol have independently been shown to be effective in patients with PAH, the authors have investigated the effect of adding oral sildenafil to long-term intravenous epoprostenol in patients with PAH as a combination therapy. In this 16-week, double-blind, placebo-controlled, and parallel-group study, 267 patients with PAH (who were under long-term intravenous epoprostenol therapy) were enrolled in this clinical trial (Simonneau et al., 2008). Patients were then randomly assigned to receive either a placebo or sildenafil (123 in the placebo group vs. 133 in the sildenafil group). Their results showed that the addition of sildenafil to long-term intravenous epoprostenol therapy significantly improved hemodynamic measurements, time to clinical worsening, quality of life, and exercise capacity, but not Borg dyspnea score (Simonneau et al., 2008). Several side effects have been reported with the combination treatment and include headache and dyspepsia. Collectively, this study indicates that sildenafil may be used in combination with epoprostenol without an apparent increase in adverse events (Simonneau et al., 2008). Of great importance, this study was conducted only in treatment-naïve patients with WHO functional class II and III PAH and includes patients with iPAH and PAH-associated with the use of anorexigen such as fenfluramine-phentermine and PAH-associated with connective tissue diseases, HIV infection, and congenital heart diseases.
In 2015, the AMBITION trial investigated the combination therapy with ambrisentan (endothelin receptor antagonist) and tadalafil (PDE-5 inhibitors) on long-term outcomes in patients with PAH (Galie et al., 2015). In this double-blind study, 500 patients with PAH were randomly assigned into three groups: 40 mg of tadalafil plus placebo (tadalafil-monotherapy group), 10 mg of ambrisentan plus placebo (ambrisentan-monotherapy group) and 10 mg of ambrisentan plus 40 mg of tadalafil (combination-therapy group). The risk of the primary endpoint of the first event of clinical failure was 50% lower among participants who received the combination therapy with ambrisentan and tadalafil than those who received only a monotherapy (Galie et al., 2015). In addition, the combination-therapy group showed lower levels of pro-brain natriuretic peptide than the monotherapy groups. Similarly, the combination-therapy group showed greater improvement in the 6-minute walk distance. Altogether, the AMBITION trial strongly supports the rationale for targeting multiple pathways in PAH by using combination therapy. In this study, the authors demonstrated that co-targeting endothelin-receptors and phosphodiesterases might be a beneficial approach (Galie et al., 2015).
Despite the current pharmacological treatments, PAH remains a fatal disease. As of today, current FDA-approved drugs showed only moderate benefits. Recent advances in the molecular understanding of PAH and cellular technology have contributed to the development of innovative strategies for treating PAH. Among them, stem cell-based therapy and genetic manipulations have open new avenues and perspectives in the treatment of PAH.
The focus on the previous FDA-approved pharmacotherapies described above attempts to overcome the imbalance between vasoactive and vasodilator mediators, and thereby restore the endothelial cell function. While the current FDA-approved drugs improve the quality of life and hemodynamic parameters, none of them offers a cure against PAH. Extensive research efforts have led to the emergence of innovative therapeutic approaches in the area of PAH. Indeed, recent preclinical studies suggested that regenerative cell-based therapies, gene therapies (Fig. 4), and epigenetic medicines may offer a new perspective in the treatment of PAH (Baliga et al., 2011; Brouwer et al., 2013; Ghofrani et al., 2009; Loisel et al., 2018).
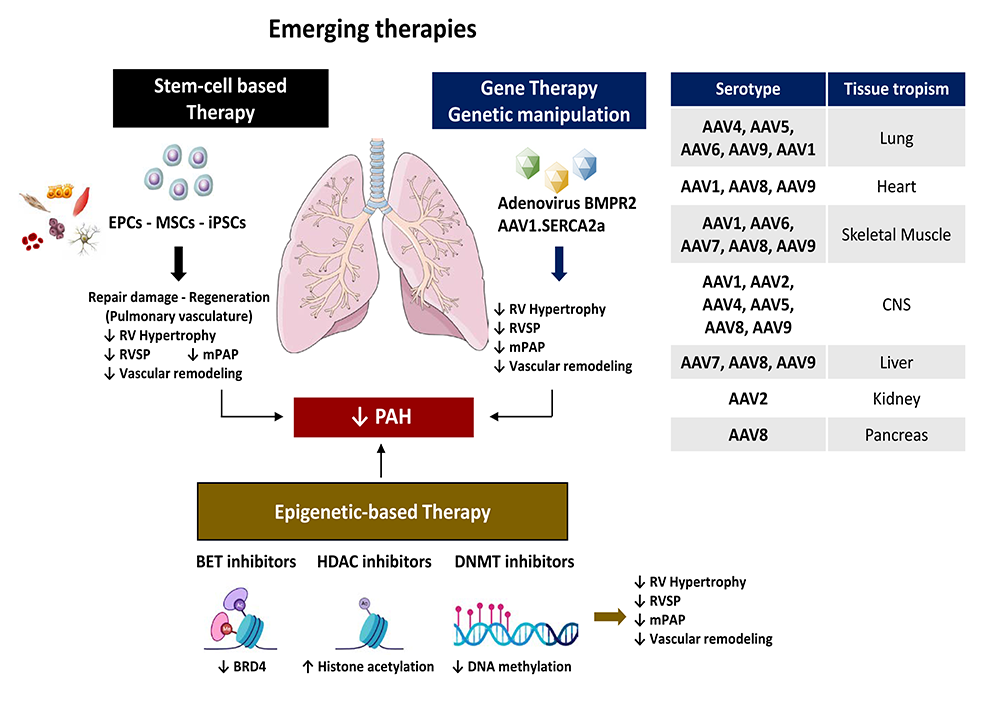 Figure 4.
Figure 4.Stem cell-based therapy, gene transfer and epigenetic therapy as promising innovative strategies for treating pulmonary hypertension. Recent preclinical studies suggested that stem cell-based therapies, gene transfer and epigenetic-based therapies may offer a new perspective in the treatment of PAH. Administration of endothelial progenitor cells (EPCs), mesenchymal stem cells (MSCs), induced pluripotent stem cells (iPSCs), intratracheal administration of inhaled-adenovirus encoding for BMPR2 or gene delivery of adeno-associated virus (AAV) serotype 1 encoding for human SERCA2a prevented and reversed the development of PH in preclinical animal models of PAH by blocking cardiac/arterial remodeling and improving hemodynamic abnormalities (RVSP, mPAP). Increasing evidence suggests that epigenetic-based therapies, such as DNMT, HDAC and BET inhibitors, may be of great therapeutic potential for treating PAH.
Increasing interest in cell-based gene therapies has contributed to demonstrating the beneficial and therapeutic role of stem cells in the treatment of PAH. Stem cells (SCs) are cells that can differentiate into other cell types, and divide in self-renewal to produce more of the same type of stem cells (Alison et al., 2002). In adult organisms, stem cells and progenitor cells act as a repair system for the body, replenishing adult tissues when injured. Nowadays, they have been isolated from several distinct sources in humans: bone marrow, adipose tissue, blood, and from umbilical cord blood just after birth. Multipotent progenitor cells and mesenchymal SCs showed strong expansion abilities in vitro, high reproducible potentials, and useful properties to differentiate (Via et al., 2012). Indeed, they can differentiate into various cell types such as vascular smooth muscle cells, muscle, bone, cartilage, or other connective tissues. Stem cell-based therapies are based on the principles of cell repair and regeneration (Mahla, 2016).
In pulmonary arterial hypertension, it applies to two pathophysiologic models- the proliferative and degenerative hypothesis. Endothelial injury and dysfunction have been shown to induce resistance to apoptosis in PAH conditions and to contribute to the formation of obliterative plexiform lesions and support the proliferative hypothesis, known as one of the main hallmarks in PAH (Sakao et al., 2009). Cell damage may induce endothelial dysfunction and loss of functional distal pulmonary vasculature. Regenerative cell therapy aims to repair and overcome endothelial injuries and dysfunction while restoring the distal pulmonary vasculature (Weiss et al., 2011). Three primary types of stem cell therapy have been described: 1- Endothelial Progenitor cell Therapy, 2-Mesenchymal stem cell therapy, and 3-Induced pluripotent stem cell therapy (Weiss et al., 2011).
5.1.1 Endothelial Progenitor cell Therapy
Endothelial progenitor cells (EPCs) were first characterized and described in 1997 as a population derived from mononuclear cells (Yoder, 2012). This finding was an important discovery and contribution to the field as it introduced the new concept that angiogenesis could occur from a distinct cell population other than angioblasts and, importantly, after embryogenesis. These cells play an essential role in post-ischemic angiogenesis (Asahara et al., 1997; Foster et al., 2014).
In preclinical studies, the administration of endothelial-like progenitor cells (derived from bone marrow) demonstrated engraftment of cells into distal pulmonary arterioles and prevented the progression of PH in the MCT-induced PH rat models. When administered 3 days after monocrotaline injection in rats, EPCs significantly decreased RV pressure. This study was the first to provide both therapeutic and preventative scope (Zhao et al., 2005). In this study, Zhao et al. demonstrated that EPCs prevented the increase of right ventricular systolic pressure. Interestingly, animals treated with EPCs transduced with human endothelial nitric oxide synthase (eNOS) showed a significant reversal of PH in the MCT-induced PH model in rats. Additionally, the delivery of early-EPCs successfully prevented the PAH onset in athymic rats in an immune system-dependent mechanism (Zhao et al., 2005). Interestingly, EPCs have been shown to be directly integrated into the distal pulmonary circulation. Moreover, EPCs exert paracrine effects through the production of pro-angiogenic cytokines and growth factors such as VEGF, stromal-derived growth factor, and Insulin-like-growth factor-1 (IGF-1) (Burchfield and Dimmeler, 2008).
Recently, Harper and collaborators have investigated the therapeutic potential of a preclinical cell therapy approach based on the upregulation of the BMPR2 signaling in PAH (Harper et al., 2019). Thus, they used bone marrow-derived endothelial-like progenitor cells isolated from the femurs of Sprague-Dawley rats. Cells were transduced with adenoviral vectors carrying the luciferase and GFP reporter gene (AdTrackLuc) or BMPR2 (AdCMVBMPR2myc). Using the MCT-induced PAH model, rats were given tail vein injections after the establishment of PH. The authors found that BMPR2-augmented rat bone marrow-derived endothelial-like progenitor cells (BMPR2-BM-ELPC) significantly increased the BMPR2 expression, and the downstream signaling in the lungs after only 24 hours. Interestingly, the treatment with BMPR2-BM-ELPC attenuated PAH, as demonstrated by the improvement in hemodynamic measurements and decreased vascular remodeling with a significant reduction in vessel thickness and muscularization (Harper et al., 2019). The authors suggested that intracellular communication via exosomes may be critical for the beneficial and protective effects on the pulmonary vasculature. The use of transduced-endothelial progenitor cells may overcome the raised concerns regarding the production of neutralizing antibodies after viral gene delivery. Furthermore, endothelial progenitor cells may be easily isolated and cultured from peripheral blood and transduced in vitro. Altogether, this study demonstrated the therapeutic efficacy of BMPR2-enhanced ELPC in a preclinical model of PAH and suggested that BMPR2-BM-ELPC may be of great value for future translational studies.
Given the success in preclinical animal models of PH, a pilot randomized control trial, including 31 patients with idiopathic PAH, was conducted in humans to assess the safety and efficacy of EPCs therapy in 2007. Patients that received SCT in addition to conventional treatments showed significant improvements in 6 minute-walk distance, mean PAP, PVR, and cardiac output at 12 weeks, without any adverse effects (Wang et al., 2007). A small pilot study was also conducted in a pediatric population with iPAH and showed improvements in hemodynamics, 6MWD, and quality of life at 12 weeks (Zhu et al., 2008). Interestingly, although these pilot studies demonstrated safety and efficacy, more extensive trials with long term follow-up remain necessary to further confirm these findings before they are incorporated into the mainstream of treatment of PH.
In 2015, the Pulmonary Hypertension and Angiogenic Cell Therapy (PHACeT) study aimed to evaluate the tolerability of culture-EPC, transiently transfected with endothelial nitric oxide synthase, in patients with PAH refractory to other PAH treatments (Granton et al., 2015). Patients were given 3 doses of endothelial nitric oxide synthase-transfected endothelial progenitor cells (eNOS-transfected EPC) directly into the right atrium on consecutive days. Their results showed that cell infusion was well tolerated, and a short-term hemodynamic improvement was reported (Granton et al., 2015). Unfortunately, no significant hemodynamic improvements were reported at 3 months despite long-term benefits in functional and quality of life, as demonstrated by an increase of the 6-minute walk distance at 1, 3, and 6 months post-delivery (Granton et al., 2015). Importantly, this study reported one serious adverse event immediately after discharge. Additional studies are of great importance to evaluate the efficacy and tolerability of eNOS-transfected EPC for treating PAH.
In line with this study, the SAPPHIRE (Study of Angiogenic Cell Therapy for Progressive Pulmonary Hypertension) study is a phase 2 clinical trial that aims to establish the efficacy and safety of repeated monthly dosing of autologous EPCs transfected with human eNOS in patients with symptomatic severe PAH on available PAH-targeted medical therapy (currently recruiting).
5.1.2 Mesenchymal stem cell therapy
Mesenchymal stem cells (MSCs) have been extensively studied and used for a variety of diseases with established safety in human trials (Lalu et al., 2012). These cells are derived from several sources, including adipose tissue, Wharton's jelly. MSCs express specific mesenchymal markers. They exhibit significant regenerative potential and play an essential role in tissue repair and angiogenesis. When injected, they have been shown to migrate to sites of injury, differentiate and promote repair (Suen et al., 2013). Surprisingly, mesenchymal cells showed low rates of tissue persistence and engraftment. Initially, the primary mechanism of the repair was thought to stem from their trophic and immunomodulatory effects mediated via the paracrine secretion of various cytokines such as IL-10, PGE2, IL-8, and VEGF (Ranganath et al., 2012).
Several advantages have been reported in the use of MSCs as stem cell transplantation. First, they are currently available, relatively easy to culture, and immune compatible, given the lack of major histocompatibility complex. Second, they showed attractive anti-inflammatory, pro-angiogenic, and anti-apoptotic properties, which make the use of MSCs an attractive treatment alternative (Foster et al., 2014). In vivo, MSCs have shown promising results in reversing and preventing the development of PAH in preclinical rodent models. In a rat-MCT PH model, intravenous delivery of MSCs after induction of PH by monocrotaline improved the right ventricular systolic pressure (RVSP), attenuated the media thickening, decreased lung collagen along with anti-inflammatory and anti-apoptotic markers (de Mendonca et al., 2017). Moreover, MSCs concomitantly improved right ventricular hypertrophy and RV ejection fraction in a monocrotaline-induced PAH rat model (Umar et al., 2009).
MSCs enrichment strategies using gene-therapy have also had success. MSCs over-expressing eNOS and Heme-oxygenase-1 (HO-1) improved RVSP and RVH in the MCT-induced PH rat model and chronic hypoxia-induced PH model respectively, compared to MSC therapy alone (Kanki-Horimoto et al., 2006; Liang et al., 2011). Enriched-MSC derived exosomes have been validated in models of hypoxic pulmonary hypertension with success. Exosomes are macrovesicles that are released from cells and have the ability to transport proteins, mRNA, etc. Lee et al. (2012) identified these exosomes as a crucial mediator for the beneficial actions of MSCs in HPH models. MSC-derived exosomes suppressed the vascular remodeling and pro-proliferative pathways in the mouse hypoxia-mediated model. Collectively, this study provides a novel avenue for cell-free therapy.
5.1.3 Induced pluripotent stem cell
Induced pluripotent stem cells (iPSCs) are adult cells that have been genetically reprogrammed from adult somatic cells to an embryonic stem cell-like state via the transduction of defined transcription factors. In 2016, Huang and colleagues have investigated the therapeutic potential of iPSCs using the MCT-induced PH rat model. Their results showed that MCT-induced PH rats that received iPSC-based therapy for prevention or reversal both showed significant improvements in hemodynamic parameters and reduced RVSP as well as RV hypertrophy (Huang et al., 2016). The histological examination further supported the beneficial effect of iPSCs-based treatments on the vascular remodeling of small pulmonary arteries. Indeed, iPSCs-treated rats demonstrated a significant decrease in media layer hypertrophy. Additionally, immunohistochemical analysis revealed that the administration of iPSCs significantly inhibited inflammation in the lungs of MCT-induced PH rats. These promising results strongly support the rationale for targeting vascular repair mechanisms in PAH in order to restore the integrity of the vascular endothelium by using an iPSCs-based therapy (Huang et al., 2016). Regenerative cell therapy has been demonstrated to improve symptoms of PAH. However, this is important to highlight that iPSCs-based therapy still requires strong optimizations and further investigation before routine clinical applications. More recently, Rabinovitch and collaborators have used induced pluripotent stem cell-derived endothelial cells human iPSC-ECs from unaffected mutation carriers, FPAH patients, and gender-matched controls to investigate the genetic variation affecting the BMPR2 gene in PAH (Gu et al., 2017). They found an increase in BMPR2 activators and a reduction in BMPR2 inhibitors that in iPSC-ECs isolated from unaffected mutation carriers. These findings correlate with the preservation of many EC functions and EC survival (Gu et al., 2017). They also demonstrated that the BMPR2 mutation is sufficient to induce EC dysfunction-associated with PAH in the absence of protective modifiers by using CRISPR/Cas9 technology.
Defective genes due to mutation or dysregulation of gene expression may contribute to disease by promoting proliferation, inducing resistance to apoptosis, damaging endothelial cells, altering calcium homeostasis (Lee and Young, 2013). Therefore, the development of safe and efficient gene-delivery systems represent a promising approach in the treatment of PAH (Fig. 4). Among these approaches, gene therapy is an experimental technique that allows the delivery of nucleic acid into a patient's cells for treating various diseases (Pfeifer and Verma, 2001). A large body of evidence has demonstrated that restoration or down-regulation of gene expression by therapeutic delivery of targeted-genes may treat and prevent PAH or inhibit the disease progression (Reynolds, 2011).
Various gene delivery strategies have been investigated and include 1-aerosol to target airway epithelium, vascular smooth muscle cells, 2- intravenous 3- intra-muscular. In the context of PAH, the intra-tracheal administration via aerosol delivery targets specifically PAECs and PASMCs, as well as the connective tissue cells/fibroblasts. Local airway delivery to the lungs remains a non-invasive route of gene administration and shows lower endonuclease activities, preventing degradation of DNA and RNA molecules (Auricchio et al., 2002; Katz et al., 2019). Different gene delivery methods can be used for local airway administration such as intranasal or oral delivery using inhalation devices, intratracheal instillation, and inhalation with or without a bronchoscope. Moreover, direct airway delivery minimizes systemic side effects and avoid liver first-pass metabolism and hepatic absorption.
5.2.1 Bone Morphogenetic Protein receptor type 2 (BMPR2)
Heritable PAH (hPAH) represents approximately 6-10% of all PAH. Genetic studies have identified more than 450 mutations heterozygous germline mutations in the Bone Morphogenetic Protein receptor type 2 (BMPR2) gene (Fessel et al., 2011). Interestingly, a mutation in the BMPR2 gene was reported in ~70% of patients with hPAH and ~20% of patients with idiopathic PAH. Besides, a moderate reduction in BMPR2 expression was also observed in patients with secondary pulmonary hypertension (Atkinson et al., 2002). Reduced expression of BMPR2 was also documented in the MCT-induced or Sugen-Hypoxia-induced PH models in rodents. Body of evidence showed that mutations in the BMPR2 gene and low expression are associated with disease pathogenesis, progression, and outcomes (Austin and Loyd, 2014). Therefore, the possibility of treating PAH via restoring BMPR2 expression and its downstream signaling are thus a rational consideration.
In 2013, Spiekerkoetter and colleagues used a transcriptional high-throughput luciferase reporter assay to screen several libraries available at the Stanford High-Throughput Bioscience Center, which include 3,756 FDA-approved drugs and bioactive compounds for the induction of the BMPR2 signaling (Spiekerkoetter et al., 2013). The authors identified the FK506 (tacrolimus) FDA-approved compound. This compound activates the BMPR2-mediated signaling and endothelial-specific gene regulation (e.g., apelin). Treatment with FK506 reversed severe established PAH in the rat MCT-induced and Sugen/hypoxia-induced PH models as well as in the conditional BMPR2 deletion mice model as demonstrated by a decrease of RVSP, PAP, and medial hypertrophy or neointima formation (Spiekerkoetter et al., 2013). Based on these findings, the same group initiated a randomized, double-blind, placebo-controlled phase 2a trial to evaluate the safety and tolerability of FK506 in stable patients with PAH (Spiekerkoetter et al., 2015). However, evidence of the efficacy of low-dose FK506 in PAH patients was inconsistent and would require further study (Spiekerkoetter et al., 2015).
Another approach to restoring the BMPR2 signaling relies on the direct delivery of BMP9, a BMPR2 ligand (Long et al., 2015). In 2015, Long et al. showed that pre-treatment of PAECs with BMP9 prevented endothelial cell apoptosis and permeability. In this study, the authors also showed that the daily administration of BMP9 by intraperitoneal injection reversed established PAH in a mouse model bearing a heterozygous knock-in of a human BMPR-II mutation, R899X (Long et al., 2015). The authors also demonstrated that daily delivery of BMP9 reversed PAH in the rat MCT-induced and Sugen/hypoxia-induced PH models as well as in the mouse Sugen-Hypoxia-induced PAH (Long et al., 2015).
In 2007, Reynolds et al. (2007) have demonstrated that targeted delivery of adenoviral vector containing the BMPRII gene attenuates hypoxic pulmonary hypertension in rodent experimental models. The authors found that BMPR2 gene delivery significantly decreased right ventricular hypertrophy, right ventricular systolic pressure, mean pulmonary artery pressure, and attenuated the hypoxia-induced vascular muscularization (Reynolds et al., 2007).
The use of adenoviral vectors remains the limiting factor of this study and may limit clinical use. Numerous studies have reported that adenovirus gene transfer is associated with an inflammatory response (Lee et al., 2017). Recent advances in gene delivery technology have led to the development of new vectors called Adeno-associated virus (AAV) (Daya and Berns, 2008; Naso et al., 2017). They remain one of the most investigated gene therapy vehicles. AAV vectors allow a selective cell and tissue-type delivery based on the sequences encoding for the Capsid. Several AAV variants are available and exhibit specific tropism properties with limited inflammatory response (Mingozzi and High, 2013). Recombinant AAV1 is currently used as a gene transfer vector in clinical trials for heart failure and in preclinical studies for treating pulmonary hypertension.
5.2.2 Sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a)
PASMCs isolated from PAH patients and animal models of PAH showed high [Ca2+]i levels. Alteration of the calcium homeostasis potentiates PASMC proliferation and migration. Therefore, it may play a critical role in vascular remodeling in PAH disease. Additionally, PASMCs isolated from IPAH patients are characterized by increased activation of the transcription factors STAT3 (signal transducer and activator of transcription-3) and NFATc2 (Nuclear factor of activated T-cells). Nuclear localization of NFATc2 and STAT3 are both associated with high [Ca2+]i and vascular remodeling in PAH. Thus, innovative approaches to restore SERCA2a expression and calcium homeostasis by AAV1-mediated gene transfer represent a new therapeutic strategy in PAH to inhibit the PASMC proliferation and vascular remodeling.
Previously, our group demonstrated that Sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) expression was significantly decreased in human lung homogenate samples from PAH patients and an experimental model of PAH such as rat monocrotaline-induced PAH (Aguero et al., 2016; Hadri et al., 2013). It has been demonstrated that SERCA2a expression was decreased in small hypertrophied pulmonary arteries in PAH. Moreover, SERCA2a overexpression was reported to inhibit Human Pulmonary Artery Endothelial (hPAEC) and smooth muscle cells (hPASMC) proliferation by restoring eNOS activation and inhibiting the NFAT/STAT3 pathway.
Furthermore, several studies have previously demonstrated that SERCA2a gene transfer via intra-tracheal delivery of aerosolized adeno-associated virus serotype 1 (AAV1) carrying the human SERCA2a gene (AAV1.SERCA2a) inhibits PAH in the MCT-induced PH rat model and chronic post-capillary pulmonary hypertension in a large animal model (Aguero et al., 2016; Hadri et al., 2013). In the MCT-induced PAH, gene transfer of SERCA2a via intratracheal delivery of AAV1.SERCA2a decreased RVSP, pulmonary artery pressure (PAP), vascular remodeling, right ventricular hypertrophy (Fulton Index), and RV fibrosis in comparison with MCT-PH rats treated with a control AAV1 carrying β-galactosidase or saline (Strauss et al., 2019). In a prevention protocol, AAV1.SERCA2a delivery attenuated adverse hemodynamic profiles as well as indices of pulmonary and cardiac remodeling in comparison with rats administered with AAV1 carrying β-galactosidase or saline. More recently, safety and long-term efficacy of AAV1.SERCA2a using nebulizer delivery has been examined in a Yukatan miniature swine model of chronic pulmonary hypertension (Watanabe et al., 2018). Similar to Yorkshire pigs, Yukatan miniature swine developed PH two months after the pulmonary vein banding surgery, as demonstrated by elevated pulmonary pressures, increased vascular resistance, and RV failure. The authors assessed the therapeutic efficacy of nebulized AAV1.SERCA2a at two months after delivery and found that nebulized AAV1.SERCA2a gene therapy significantly decreased pulmonary vascular medial thickness, pulmonary vascular resistance, and increased long-term survival compared to control animals (Watanabe et al., 2018).
Over the past decade, accumulated data suggested that epigenetics may play a major role in the setting of PAH (Bisserier et al., 2020; Cheng et al., 2019). Epigenetics is defined as a heritable change to the chromatin resulting in a shift in gene expression without altering the DNA sequence (Weinhold, 2006). These changes are mediated, in part, through specific epigenetic modifications called epigenetic marks and include methylation, acetylation, and phosphorylation of the DNA or histone proteins. These epigenetic marks affect the condensation of chromatin, the accessibility of transcription factors, transcriptional activation, and gene expression (Weinhold, 2006).
5.3.1 DNA methyltransferase (DNMT) inhibitors
For example, DNA methylation has been shown to be associated with repression and silencing of gene expression. Interestingly, hypermethylation of the BMPR2 promoter has been reported in peripheral blood mononuclear cells (PBMCs) from hPAH patients by genomic bisulfite sequencing (Liu, D. et al., 2017). Previous studies have reported that reduced BMPR2 expression in lung tissue potentiates cell proliferation and may be sufficient to induce PAH. In 2010, Archer and collaborators found that the level of the mitochondrial superoxide dismutase 2 (SOD2) was significantly decreased in fawn-hooded rats (FHR), known to develop PAH spontaneously (Archer et al., 2010). As no change in the DNA sequence was found, the authors undertook bisulfite sequencing of FHR-derived PASMCs and identified hypermethylation of the CpG islands within the SOD2 promoter region. Pharmacological inhibition of DNMTs by 5-aza-2′-deoxycytidine restored SOD2 expression, inhibited cell proliferation and apoptosis resistance in FHR-derived PASMC (Archer et al., 2010). Collectively, this study suggested that targeting DNA methylation could be an attractive therapeutic option to restore SOD2 expression and inhibit PAH.
5.3.2 Histone deacetylase (HDAC) inhibitors
Histone deacetylase (HDAC) are a class of enzyme that removes the acetyl group from histone proteins, affecting the chromatin compacting and accessibility of transcription factors to DNA (Seto and Yoshida, 2014). This alters the transcriptional machinery and is usually associated with transcriptional repression. Preliminary studies revealed that HDAC1, HDAC5, and HDAC5 were upregulated in PAH patients and experimental models of PH (Zhao et al., 2012). HDAC inhibitors (HDACi) have been FDA-approved for the treatment of different types of cancer. For example, SAHA (also known as Vorinostat) has been approved for the treatment of cutaneous T-cell Lymphoma (CTCL) (Eckschlager et al., 2017). FK228 (also known as Romidepsin) has been approved for treating CTCL in combination with a systemic therapy (Eckschlager et al., 2017). Belinostat (also known as PXD101) has been approved against peripheral T-cell lymphoma (PTCL) (Eckschlager et al., 2017).
In the MCT-induced and pulmonary artery banding-induced PH rat model (PAB), HDAC inhibitors (Valporic Acid) demonstrated beneficial effects by significantly inhibiting the RV remodeling and improving the RV systolic function (Cho et al., 2010). Similarly, the MC1568 compound (a selective inhibitor of class IIa HDACs) inhibited MCT-induced PH and Sugen Hypoxia-induced PH in rats by restoring the expression of the myocyte enhancer factor-2 (MEF2) in lungs (Kim et al., 2015). Altogether, these independent studies further confirmed the therapeutic potential of targeting histone acetylation using HDAC inhibitors in PAH to inhibit the vascular remodeling, RV hypertrophy and improve the cardiac function.
5.3.3 Bromodomain and Extra-Terminal motif (BET) inhibitors
While HDAC proteins regulate the acetylation level, histones acetylation marks are read by bromodomain-containing proteins (BRD), such as the bromodomain and extra-terminal domain (BET) proteins (Muller et al., 2011). The BET family includes 3 members that are ubiquitously expressed: BRD2, BRD3, BRD4, and the bromodomain testis-specific protein (BRDT) proteins. Of the BET family proteins, BRD4 remains the most investigated member. It is an epigenetic regulator that recognizes the acetylated histones H3 and H4 via its tandem N-terminal bromodomains (Muller et al., 2011). It acts as a transcriptional regulator via its C-terminal bromodomains. By interacting with the transcription elongation factor complex (pTEFb), BRD4 regulates the transcriptional elongation mediated by RNA polymerase II (RNA Pol II) in a phosphorylation-dependent mechanism. It controls the expression of essential genes involved in various biological functions such as apoptosis, proliferation, migration, differentiation, cell cycle, and inflammation (Liu, Z. et al., 2017). Thus, substantial efforts have been dedicated to the identification and development of BRD4 inhibitors for treating cancer.
In 2015, Meloche et al. identified a significant increase in BRD4 expression in lung biopsies, distal pulmonary arteries, and PASMCs from PAH patients compared to controls (Meloche et al., 2015). Using a loss of function approach, the authors found that the inhibition of miR-204 increased the BRD4 mRNA levels, suggesting a possible interaction with miR-204. Similarly, the restoration of miR-204 in PAH-PASMCs, using a synthetic miR-204 mimic, inhibited BRD4 expression. In this study, the authors showed that JQ1, a pharmacological inhibitor of BRD4, decreased the NFATc2 and Bcl-2 mRNA levels as well as the nuclear translocation of NFATc2 (Meloche et al., 2015). Similar results were obtained after BRD4 knockdown using a specific siRNA against BRD4. They also demonstrated that the BRD4 inhibition triggers the proliferation/apoptosis imbalance in PAH-PASMCs. In vivo, BRD4 inhibition, by nebulization of either siRNA (siBRD4) or a clinically available inhibitor (JQ1), improved the hemodynamic measurements (mPAP, RVSP, cardiac output) and inhibited the RV hypertrophy in the Sugen/hypoxia model of PH. Histological analysis revealed that the BRD4 inhibition restored the proliferation/apoptosis balance within the vascular wall, which is associated with a reduction in vascular remodeling (Meloche et al., 2015). Finally, recent results from a multicenter randomized preclinical trial performed in three different labs independently demonstrated and confirmed the therapeutic potential of the inhibitor RVX208 in several rodent models of PAH (Van der Feen et al., 2019). These results provided the basis for the launch of a promising phase 2 clinical trial in PAH patients (Pullamsetti and de Jesus Perez, 2019).
Despite the current approved-therapies, PAH remains a progressive and fatal disease. Indeed, the available and FDA-approved pharmaceutical therapies target mainly three major pathways: the prostacyclin pathway, the endothelin pathway, and the PDE-5 pathway. All these drugs (epoprostenol, treprostinil, iloprost, bosentan, ambrisentan, sildenafil, tadalafil, riociguat) aim to overcome the imbalance between vasoactive and vasodilator mediators, restore the endothelial cell function and decrease the vascular remodeling. Despite the beneficial effects on survival, disease progression, and quality of life, the current therapies do not cure PAH, which remains a fatal disease. Recent technological advances and a better understanding of the underlying molecular mechanisms in PAH have led to the development of new tools and strategies for the treatment of PAH. Among them, stem cell-based therapy, aerosolized gene therapy and epigenetic medicines have shown promising results for the treatment of PAH in preclinical studies and require further investigation before consideration for clinical translation.
MB, NP, and LH contributed to writing and editing the manuscript. All authors have approved the final version.
This study was supported by the NIH R01 HL133554 (to LH) and AHA Innovative Project Award 18IPA34170321 (to LH), NIH 5T32HL007824-22 and Cardiovascular Medical Research and Education Fund (CMREF) (to MB).
Not applicable. Only the authors listed on the manuscript contributed towards the article.
The authors have declared that no conflict of interest exists.