
Academic Editor: Brian Tomlinson
Histone deacetylase (HDAC) inhibitors have shown cardioprotective or renoprotective effects in various animal models. Our study proposed that the HDAC inhibitor, mocetinostat, regulates cardiac remodelling and renin-angiotensin system (RAS) activity in rats with transverse aortic constriction (TAC)-induced pressure overload cardiac hypertrophy. Cardiac remodelling was evaluated using echocardiography. Cardiac hypertrophy was visualized with haematoxylin and eosin staining, and related gene (Nppa and Nppb) expression was quantified by quantitative real-time polymerase chain reaction (qRT-PCR). Cardiac and renal fibrosis were visualized with picrosirius red and trichrome staining, respectively. Fibrosis related gene (Collagen-1, Collagen-3, Ctgf, and Fibronectin) expression was determined by qRT-PCR. Serum concentrations of RAS components (renin, angiotensin II, and aldosterone) were quantified by enzyme-linked immunosorbent assay and related gene (Renin and Agtr1) expression was determined by qRT-PCR. TAC-induced pressure overload cardiac hypertrophy, which mimics hypertensive heart disease, increased cardiac remodelling, cardiac hypertrophy, and fibrosis in our rat models. Upon treatment with mocetinostat, there was a significant regression in cardiac remodelling, cardiac hypertrophy, and fibrosis in TAC rats. Additionally, pressure overload-induced renal fibrosis and activity of RAS-related components were increased in TAC rats, and were decreased on treatment with mocetinostat. The present study indicates that mocetinostat, an HDAC inhibitor, has cardiorenal protective effects in rats with TAC-induced pressure overload cardiac hypertrophy and offers a promising therapeutic agent for hypertension-related diseases.
Hypertension (HTN) is a major modifiable risk factor for cardiovascular disease. The left ventricle is the primary target of HTN related end-organ damage. In addition to being a marker of HTN, cardiac remodelling of left ventricle occur initially as compensatory responses to increased systemic pressure overload, which modifies the performance and changes the geometry of the heart. Blood pressure control with lifestyle changes and anti-hypertensive agents prevents and regresses left ventricular hypertrophy (LVH) [1, 2]. Furthermore, the relationship between HTN and renal disease is well-known. Although progression to end-stage renal disease is relatively low in uncomplicated essential HTN, mild renal dysfunction could occur, such as a subclinical end-organ damage in untreated patients with primary HTN. Thus, the presence of LVH could represent a relationship between renal failure and cardiovascular diseases in all stages of renal failure, starting from the earliest stage [3, 4].
Recent studies have suggested that the left ventricle remodelling begins much earlier than expected and is seen in normotensive patients with mild renal dysfunction when the glomerular filtration rate is still normal. Strategies to reduce LVH include lowering the blood pressure and reducing hypervolemia [5]. Numerous medications, such as angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, beta-blockers, calcium channel blockers, and diuretics, have been shown to regress the blood pressure to normal levels. Nevertheless, numerous reports have raised concerns that these current existing medications do not reverse the remodelled heart to the previous normal physiology, which highlights the need of discovering new agents that can protect or reverse cardiac remodelling in conditions of hemodynamic pressure overload [1, 2, 6].
Various studies have shown that histone deacetylase (HDAC) inhibitors are effective in the treatment of cancer, autoimmune diseases, inflammatory diseases, interstitial fibrosis, cardiovascular diseases, and renal diseases [7, 8]. Our previous studies reported that valproic acid has cardiac and vascular protective effects [2] and mocetinostat attenuates aortic remodelling [9] in pressure overload-induced cardiac hypertrophy. Other reports have also reported the effects of numerous HDAC inhibitors in various animal models of cardiovascular or renal disease. Valproic acid [10, 11] and CG200475 [12] attenuate hypertrophy and fibrosis of the heart in DOCA-salt-induced hypertensive rats. Trichostatin A, valproic acid, and SK-7041 prevent hypertrophy of the heart in animal models of angiotensin II-infused cardiac hypertrophy [13]. Valproic acid ameliorates the angiotensinogen II production in the kidney in high-fat diet-induced HTN model [7]. Trichostatin A [14] and CG200745 [15] attenuate renal fibrosis in unilateral ureteral obstruction mouse model.
Mocetinostat (MGCD0103), is a chemical synthesised aminobenzamide class I (HDAC1, HDAC2, and HDAC3) or IV (HDAC11) HDAC inhibitor which widely inhibits HDAC1 and is oral route administeration. Studies have reported its anti-fibrotic capacities [9, 16, 17]. The transverse aortic constriction (TAC) animal model represents pressure overload cardiac hypertrophy by introducing LVH via the pressure of systemic HTN [18]. Our study proposed that the HDAC inhibitor, mocetinostat regulates cardiac remodelling and renin-angiotensin system (RAS) activity in rats with TAC-induced pressure overload cardiac hypertrophy.
Sprague Dawley male rats (Samtako, Osan, South Korea), 11-weeks-old, were housed
in a 12 h light/12 h dark cycle and at constant temperature (22
The blood pressure was measured for five weeks using a non-invasive method as
follows. Briefly, the hotplate was preheated (10 min, 35
Cardiac remodelling was estimated by transthoracic echocardiography with 12 MHz sector array transducer used for paediatric/neonatal cardiology (Philips, Amsterdam, Netherlands). Isoflurane (1–3%, Hana Pharm. Co. Ltd., Seoul, South Korea) gas inhalation was used to anesthetize the rats during echocardiography examination. Through parasternal long axis view, M-mode images were used to evaluate the heart wall thickness at the end of the systole phase.
Heart and kidney tissues were processed for histology and images of the slices were digitalized using the protocol reported in our previous studies [2, 9]. The tissues were stained with haematoxylin and eosin, picrosirius red, or trichrome stain. The cross-sectional areas of the heart tissue were calculated by using the ImageJ software (http://rsbweb.nih.gov; National Institutes of Health, Bethesda, Maryland, USA), an automatic quantification program.
To quantify the level of natriuretic peptide A (Nppa) and natriuretic peptide B (Nppb), the markers for hypertrophy of the heart, and Collagen-1, Collagen-3, Ctgf, and Fibronectin, markers for fibrosis of the heart and kidney, quantitative real-time polymerase chain reaction (qRT-PCR) was performed. qRT-PCR was performed as previously reported [2, 9]. Sequence of primer set are listed in Table 1.
| Gene (Accession No.) | Primer sequence (5 | |
| Nppa (NM_012612) | F: | ATCTGATGGATTTCAAGAACC |
| R: | CTCTGAGACGGGTTGACTTC | |
| Nppb (NM_031545) | F: | ACAATCCACGATGCAGAAGCT |
| R: | GGGCCTTGGTCCTTTGAGA | |
| Collagen-1 (NM_053304) | F: | GTCGAGGGCCAAGACGAAG |
| R: | CAGATCACGTCATCGCACAAC | |
| Collagen-3 (NM_032085) | F: | CTGGTCCTGTTGGTCCATCT |
| R: | ACCTTTGTCACCTCGTGGAC | |
| Ctgf (NM_022266) | F: | TCCCGTTAGCCTCGCCTTGG |
| R: | CGGTACACGGACCCACCGAA | |
| Fibronectin (NM_019143) | F: | AGCAAATCGTGCAGCCTCCG |
| R: | CCCCCTTCATGGCAGCGATT | |
| Renin (NM_012642) | F: | GTAACTGTGGGTGGAATCATTGTG |
| R: | TGGGAGAGAATGTGGTCGAAGA | |
| Agtr1 (NM_030985.4) | F: | GGAGAGGATTCGTGGCTTGAG |
| R: | CTTTCTGGGAGGGTTGTGTGAT | |
| Gapdh (NM_017008) | F: | TGCACCACCAACTGCTTAG |
| R: | GATGCAGGGATGATGTTC | |
After sacrificing the rats, the collected serum sample was stored at –80
The data are expressed as mean
The blood pressure was estimated and reported for five weeks. TAC group
exhibited a significant increase in systolic blood pressure of nearly 45 mmHg
(p
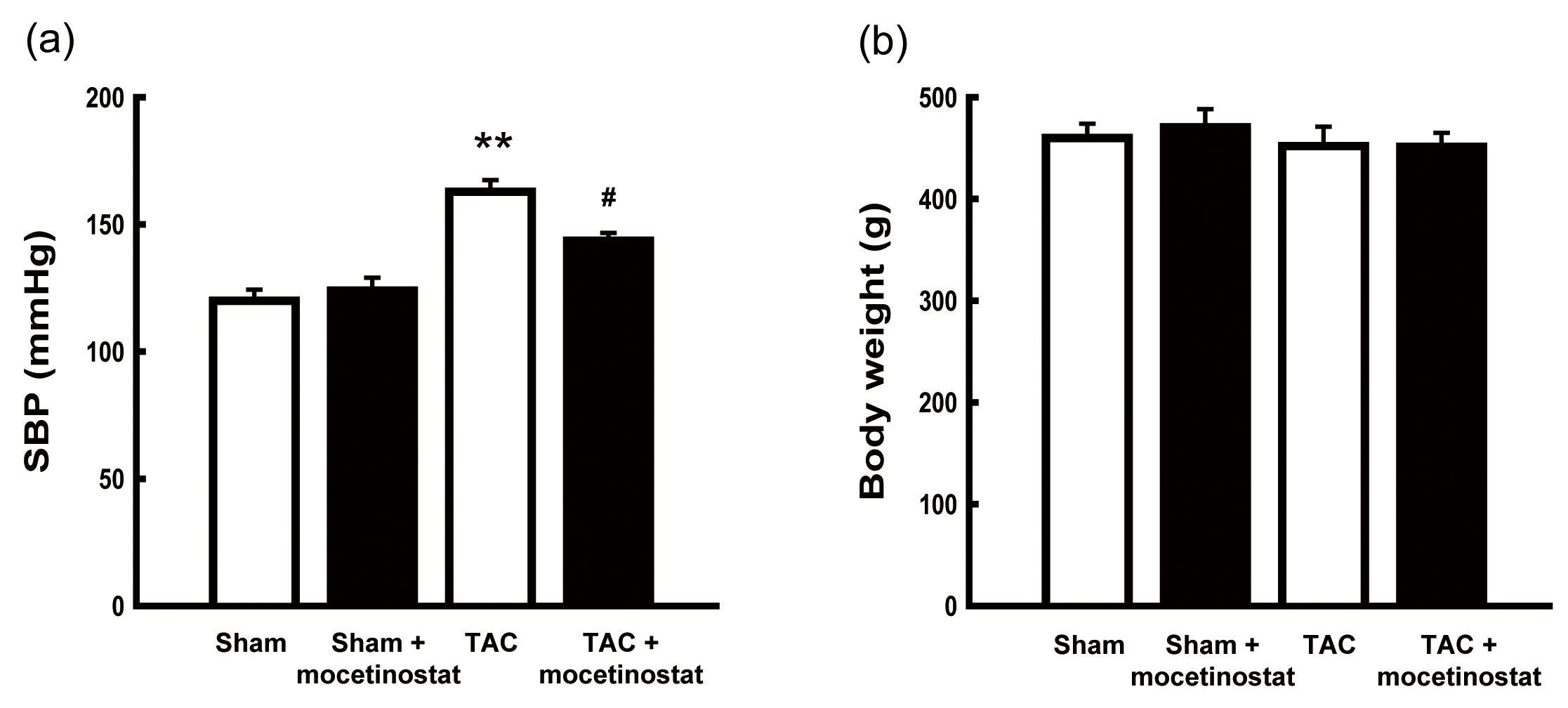 Fig. 1.
Fig. 1.Effect of mocetinostat on physical measurements in rats after
transverse aortic constriction (TAC)-induced pressure overload. (a) Systolic
blood pressure (SBP) was estimated for five weeks. Treatment with mocetinostat
reduced the blood pressure induced by TAC. (b) Body weight was estimated for four
weeks. The body weight was not affected in any of the four groups.
#, p
Echocardiography results demonstrated that mocetinostat regulated cardiac
remodelling in the left ventricle. The heart wall thickness of interventricular
septum (IVS, p
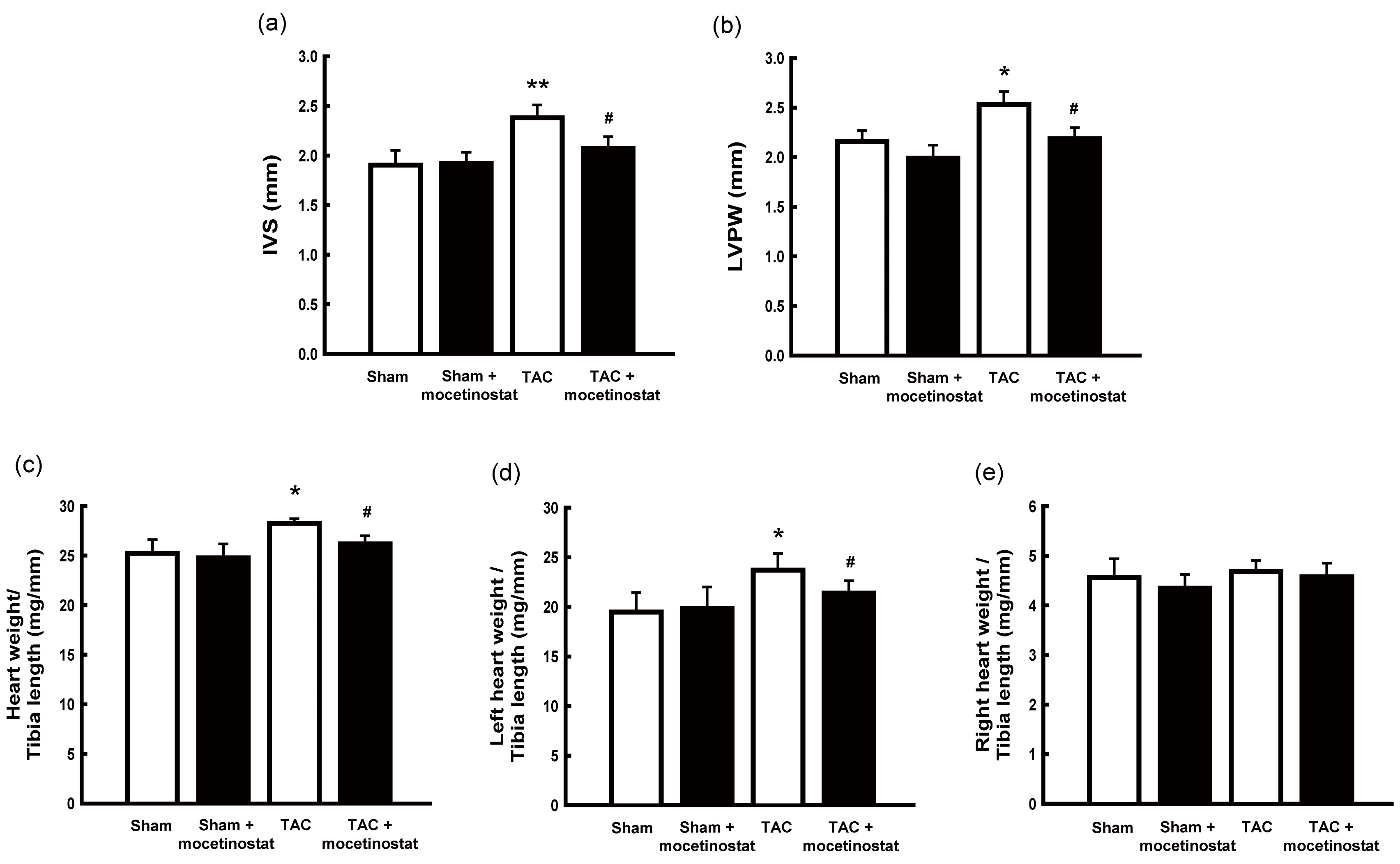 Fig. 2.
Fig. 2.Effect of mocetinostat on cardiac remodelling in rats after
TAC-induced pressure overload. (a,b) Left ventricular remodelling were evaluated
by echocardiography. The thickness of interventricular septum (IVS, a) and left
ventricular posterior wall (LVPW, b) were estimated. Treatment of mocetinostat
suppressed TAC-induced cardiac remodelling. Cardiac hypertrophy was estimated by
the weight/tibia length ratio of the heart. The weight/tibia length ratios of the
heart (c) and left heart (d) were increased in TAC group compared with those in
sham group. The administration with mocetinostat restored the weight/tibia length
ratios of the heart (c) and left heart (d). Mocetinostat administration did not
affect the weight/tibia length ratios of the right heart (e).
*, p
The weight/tibia length ratio of the heart revealed the changes related to
hypertrophy-induced cardiac mass. The weight of the heart was remarkably
increased (p
To demonstrate histological LVH in TAC group, the sections were stained with
haematoxylin and eosin. The heart wall thickness of the left ventricle was
increased in TAC group compared with sham group, and mocetinostat treatment
moderated the heart wall thickness of the left ventricle in TAC group (Fig. 3a).
Additionally, the hypertrophy of cardiomyocytes (Fig. 3b) was considerably
increased (p
The expression of Nppa and Nppb, markers of hypertrophy of the
heart, were measured using qRT-PCR. The mRNA level of Nppa (p
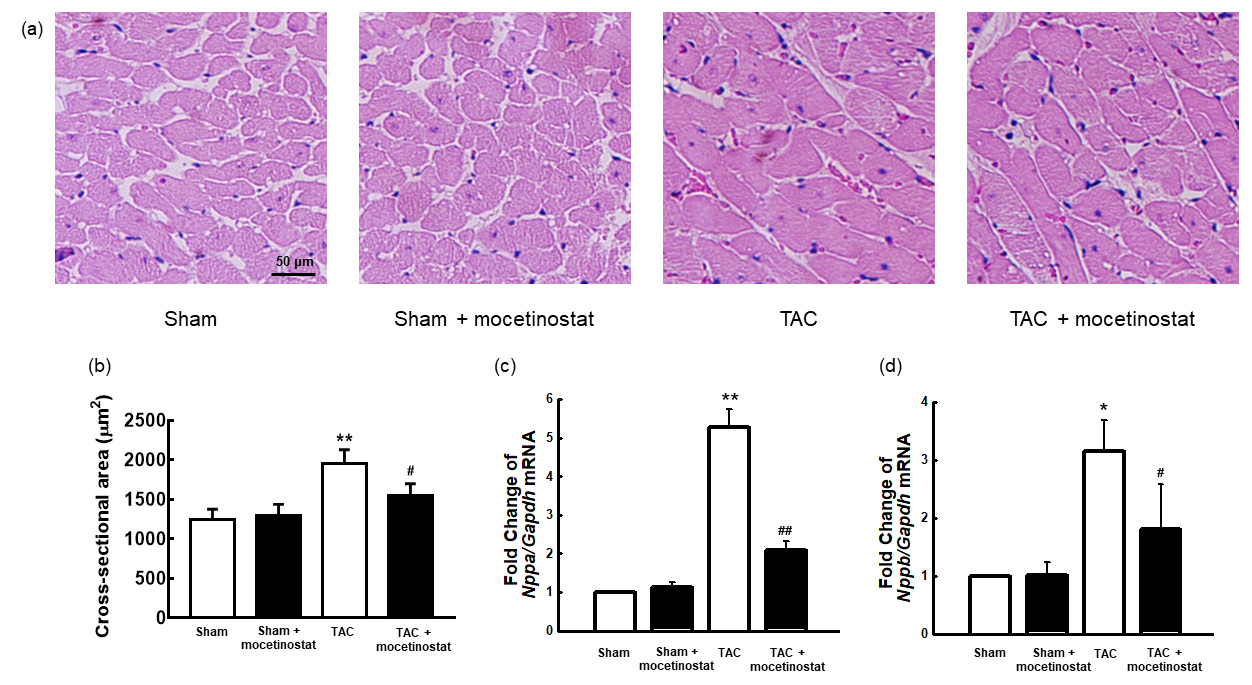 Fig. 3.
Fig. 3.Effect of mocetinostat on cardiac hypertrophy in rats after
TAC-induced pressure overload. Histological observation of the hearts was
acheived by haematoxylin and eosin staining. The hypertrophy of the
cardiomyocytes (a,b) were increased in TAC group compared with those in sham
group, which were moderated by mocetinostat administration. Scale bar shows 50
*, p
Picrosirius red staining revealed heart fibrosis and collagen accumulation in TAC group. Collagen accumulation was increased in TAC group compared with sham group, and mocetinostat administration moderated collagen accumulation in TAC plus mocetinostat group (Fig. 4a).
The mRNA level of Collagen-1, Collagen-3, Ctgf, and
Fibronectin, markers for the fibrosis of the heart, was remarkably
increased (all p
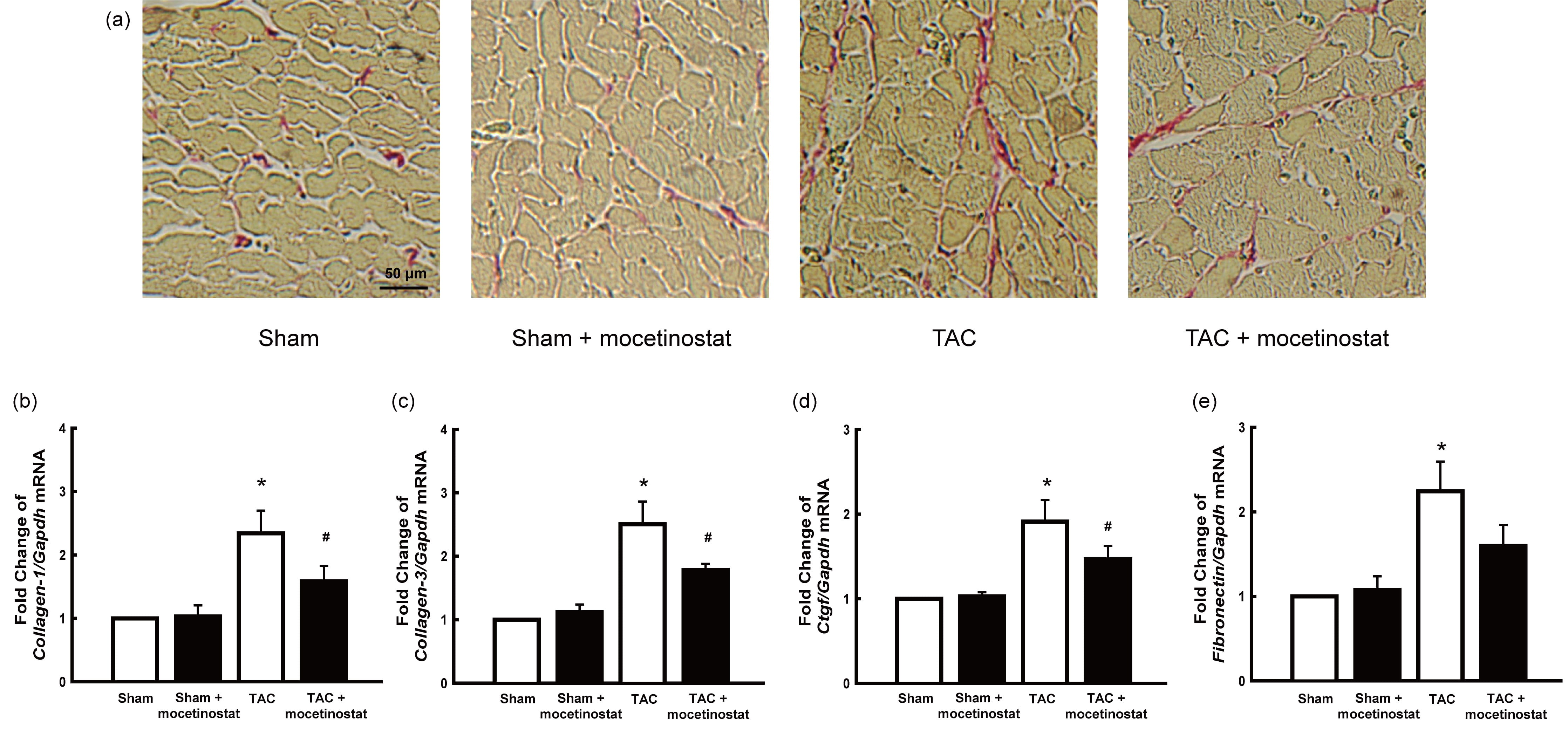 Fig. 4.
Fig. 4.Effect of mocetinostat on cardiac fibrosis in rats after
TAC-induced pressure overload. Histological observation of the hearts was
acheived by picrosirius red staining. TAC group presented increase of collagen
accumulation and fibrosis of the myocardium (a, red stain) when compared with
those in sham group. Mocetinostat administration moderated cardiac fibrosis in
TAC plus mocetinostat group. Scale bar shows 50
*, p
To analyse the kidney morphology, we performed haematoxylin and eosin (Fig. 5a) and trichrome staining (Fig. 5b). Collagen accumulation was increased in TAC group compared with sham group, and mocetinostat treatment moderated collagen accumulation in TAC plus mocetinostat group (Fig. 5b).
The mRNA level of Collagen-1, Collagen-3, Ctgf, and
Fibronectin, markers for the fibrosis of the kidney, was remarkably
increased (all p
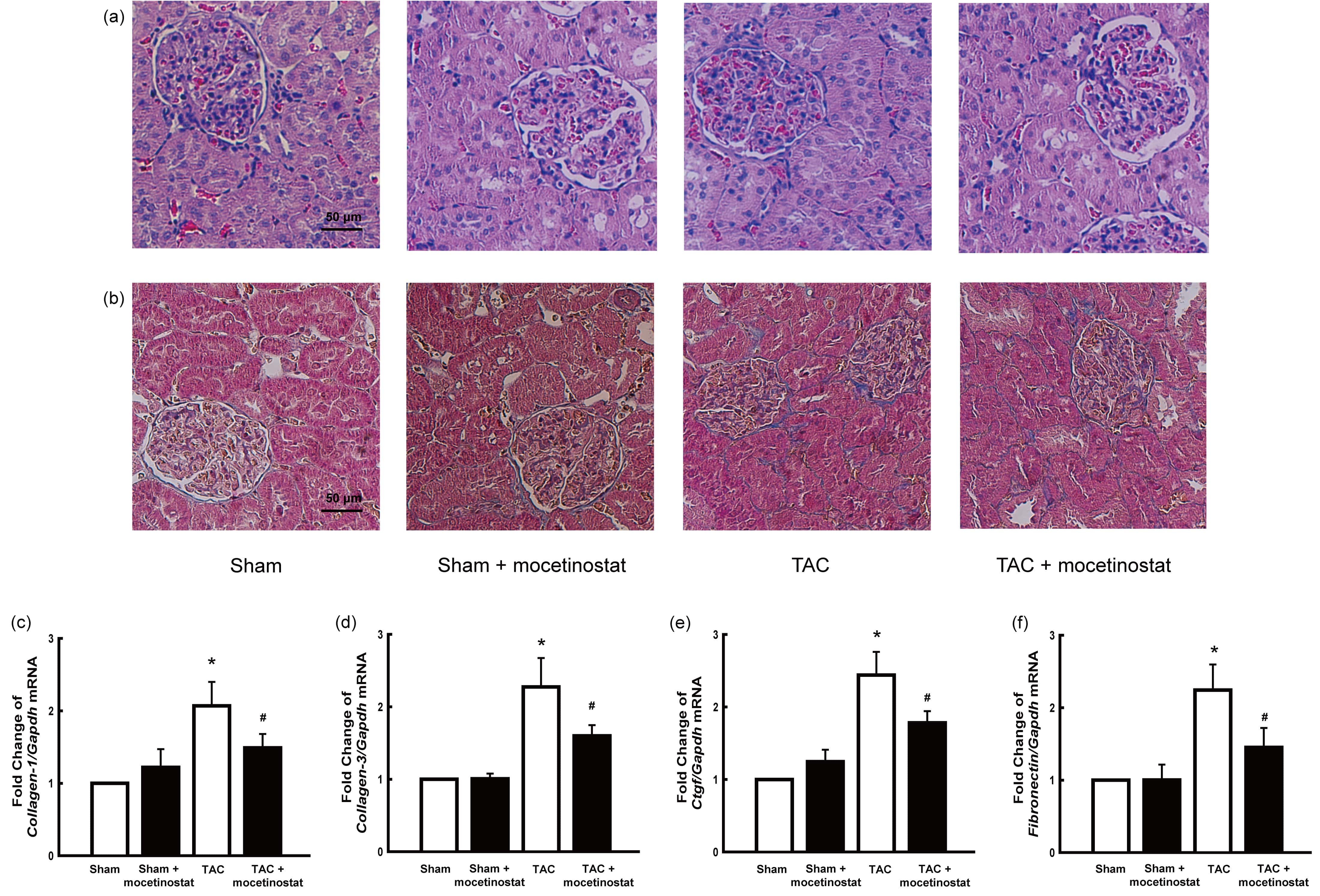 Fig. 5.
Fig. 5.Effect of mocetinostat on renal fibrosis in rats after
TAC-induced pressure overload. Histological observation of the kidneys was
achieved using haematoxylin and eosin (a), and trichrome (b) staining. TAC group
presented increase of collagen accumulation and fibrosis (b, blue stain) when
compared with those in sham group. Mocetinostat administration moderated renal
fibrosis in TAC plus mocetinostat group. Scale bars show 50
*, p
The expression of the RAS components was analysed by measuring the serum
concentrations of renin, angiotensin II, and aldosterone. Serum concentrations of
renin (p
qRT-PCR of the kidney tissue was performed to identify the activity of renin and
angiotensin II receptors, and their possible regulation by mocetinostat.
Renin (Fig. 6d) and Agtr1 (Fig. 6e) mRNA expression was
significantly higher (p
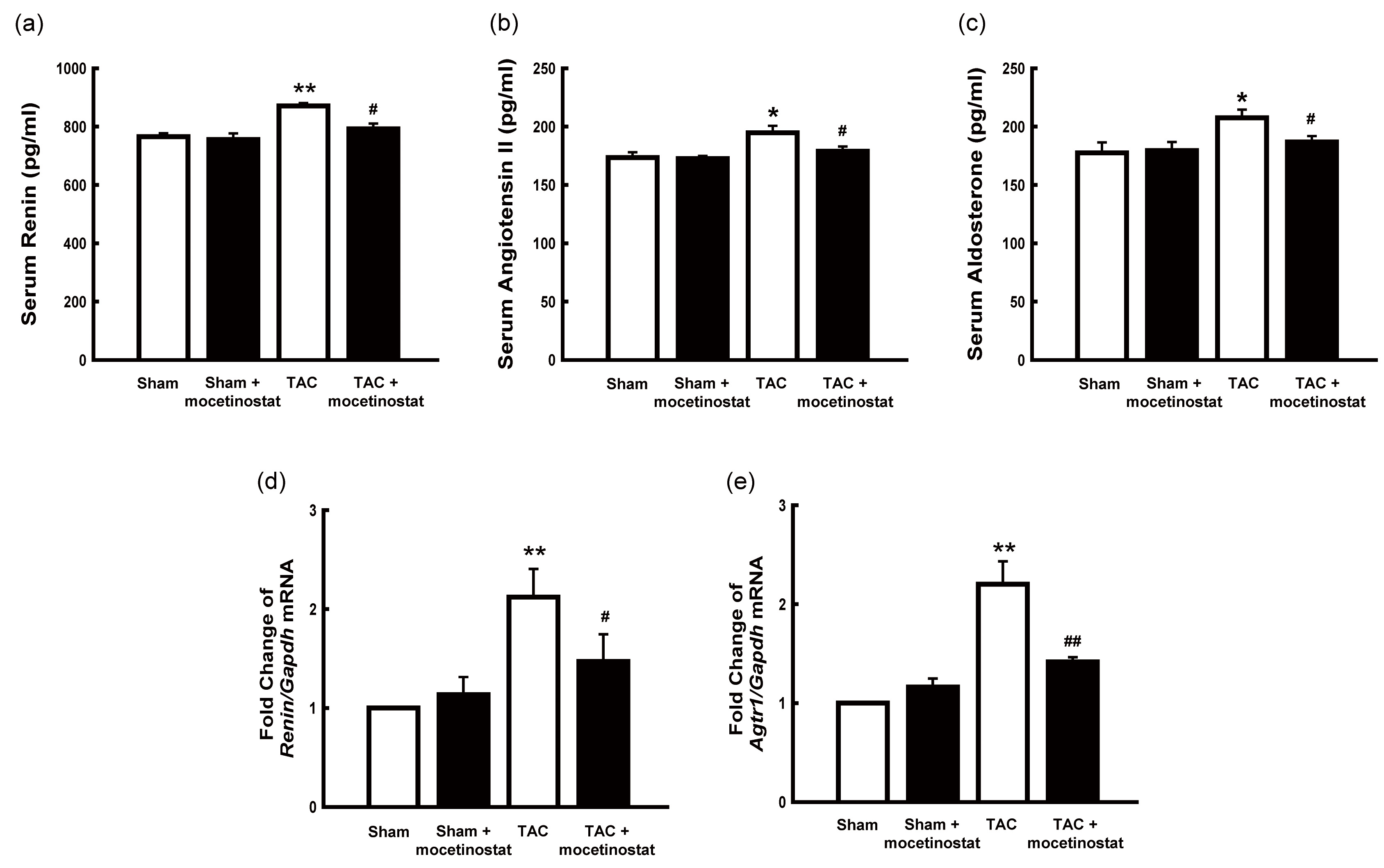 Fig. 6.
Fig. 6.Effect of mocetinostat on the renin-angiotensin system in rats
after TAC-induced pressure overload. Serum concentration of renin, angiotensin
II, and aldosterone were measured. Renin (a), angiotensin II (b), and aldosterone
(c) were significantly increased in TAC group compared with sham group, and
mocetinostat considerably regulated these concentrations in TAC group. TAC group
had increased Renin (d) and Agtr1 (e) mRNA expression, which
decreased with mocetinostat administration.
*, p
In the current experiment, we presented that mocetinostat regulates cardiac remodelling and RAS activity in rats with TAC-induced pressure overload cardiac hypertrophy. The results demonstrated that mocetinostat administration regulated cardiac hypertrophy and fibrosis and correspondingly attenuated cardiac remodelling. It also regulated renal fibrosis and RAS activation during pressure overload.
Many reports have shown that HDAC inhibitors have cardioprotective or renoprotective effects in various animal models. The TAC-induced cardiac hypertrophic animal model mimics HTN or pressure overload in patients. HTN or pressure overload conditions, which cause cardiac hypertrophy, fibrosis, and remodelling, not only influences the heart, but also the kidney, resulting in mild renal dysfunction with normal serum creatine levels [3, 4]. Our study aimed to investigate the effect of HDAC inhibitors on the heart and kidney in a pressure overload animal model in order to identify a novel therapeutic agent to treat patients with HTN.
Hypertrophy of the heart is induced by systemic pressure overload in rat’s aorta via TAC; moreover, the gradually increasing pressure on the aorta may induce LVH as a consequential response to hemodynamic pressure and cause HTN [2, 9]. Our study was performed for four weeks, which was a relatively short period to develop left ventricular dysfunction or heart failure but long enough to induce LVH. LVH is characterised by thickened IVS and LVPW, increased left heart weight, cardiomyocyte hypertrophy, and increased gene expression of hypertrophy markers, such as Nppa and Nppb. Similar to previous studies [2, 9, 11, 12, 13], the above results also confirmed the cardioprotective effect of mocetinostat in pressure overload-induced LVH. In our study, mocetinostat treatment regressed IVS and LVPW, decreased left heart weight and cardiomyocyte size, and attenuated the gene expression of Nppa and Nppb (Figs. 2,3). As mocetinostat moderates the hypertrophy of the heart, hemodynamic pressure overload may reduce, and correspondingly decreases the blood pressure. This not only effects the systemic HTN but also the pulmonary HTN. Timothy A. McKinsey et al. [19] reported that mocetinostat possibly reduces the pulmonary artery pressure or the pulmonary HTN. In this study, rats were housed in hypobaric chambers to induce hypoxia, right ventricle hypertrophy, and pulmonary HTN. We assume that mocetinostat moderates the hypertrophy of the heart, either the left or right ventricle, depending on the animal model, and correspondingly decreases systemic or pulmonary HTN, respectively.
Fibrosis of the heart is characterised by collagen deposition and myocardial fibrosis. Fibrosis of the heart consequently increases with hypertrophy of the heart, resulting in left ventricular stiffness and cardiac remodelling [2]. HDAC inhibition diminishes cardiac myofibroblast activation and reduces cardiac fibrosis in a congestive heart failure animal model [20] and in rats with angiotensin II-infused heart dysfunction [6]. In the current study, mocetinostat attenuated histological fibrosis and expression of Collagen-1, Collagen-3, Ctgf, and Fibronectin mRNA (Fig. 4). Moreover, mocetinostat treatment suppressed left ventricular stiffness.
Chronic HTN, due to high blood pressure may induce mechanical shear stress along the aorta, heart, and kidneys and oxidative stress with chronic inflammation, and reparative mechanisms of these lead to end-organ damage, mainly fibrosis [21]. Kidney damage occurs along the entire course of HTN, ranging from benign to malignant form of nephropathy, relying on an individual’s susceptibility, level of HTN, type of aetiology, and underlying disease [22]. However, long-term HTN eventually results renal interstitial fibrosis, which activates renal fibroblasts and generates excessive deposition of extracellular matrix proteins. Moreover, tubular hypertrophy, oxidative stress, activation of the RAS, collagen turnover, chronic inflammation, and secretion of vasoactive substances synergistically contribute to the hypertensive renal fibrosis [23]. Several studies have reported the therapeutic capacity of HDAC inhibitors in various renal disease animal models. CG200745 attenuates renal fibrosis in a mouse model of obstructive kidney disease [15] and high-fat diet-induced HTN [7], and valproic acid ameliorates renal fibrosis in diabetic rats [24]. We treated TAC-induced pressure overload HTN rats with mocetinostat, an HDAC inhibitor. Mocetinostat attenuated histological fibrotic changes and the mRNA level of Collagen-1, Collagen-3, Ctgf, and Fibronectin, which were increased via pressure overload cardiac hypertrophy (Fig. 5). Classically, the kidney has an auto-regulatory mechanism (glomerular afferent arteriole contraction and tubuloglomerular feedback) to protect itself from sudden increase in blood pressure [22]. RAS is the most important and well-known hormonal system in controlling the blood pressure. HTN occurs by increased resistance of blood vessels and relates to the structural changes of the blood vessels. Angiotensin II is a vasoconstrictor hormone, which induces vascular remodelling and inflammations in the arteries in the heart and kidney [25, 26]. Components of the RAS importantly function to control the blood pressure, but they are also associated with the onset of HTN. Due to these functions, there have been attempts to identify new drugs targeting RAS components [21]. We investigated the serum concentration (renin, angiotensin II, and aldosterone) and mRNA expression (Renin and Agtr1) of the RAS components. In our study, mocetinostat treatment reduced the serum concentration of renin, angiotensin II, and aldosterone, suggesting that the HDAC inhibitor suppressed the RAS and eventually decreased the blood pressure (Fig. 6).
The current study has a few limitations. First, the detailed molecular signalling pathway of mocetinostat in regulating cardiac remodelling and RAS activity need to be further explored in the further investigation. Second, the clinical importance of mocetinostat must be studied in patients with HTN-related diseases.
In conclusion, the present study revealed that mocetinostat, an HDAC inhibitor, restores left ventricular remodelling, attenuates serum RAS components, and regulates the increased mRNA levels associated with cardiac hypertrophy (Nppa and Nppb), fibrosis (Collagen-1, Collagen-3, Ctgf, and Fibronectin), and RAS components (Renin and Agtr1). Therefore, mocetinostat has cardiorenal protective effects in TAC-induced pressure overload cardiac hypertrophy rats.
HDAC, histone deacetylase; HTN, hypertension; IVS, interventricular septum; LVH, left ventricular hypertrophy; LVPW, left ventricular posterior wall; RAS, renin-angiotensin system; TAC, transverse aortic constriction.
GJK, HJ, EL and SWC designed the experiment. HJ and EL conducted the experiment and collected the data. GJK, HJ and EL analysed the data and wrote the manuscript. HJ, EL and SWC edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Institutional Review Board of Kyungpook National University approved the animal experiment (Approval number: KNU2018-79). The animal experiment was conducted as per the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.