
Academic Editor: Ichiro Wakabayashi
Although many endocrine diseases can be associated with acquired cardiomyopathy and heart failure, conditions except hypothyroidism, hyperthyroidism, phaeochromocytoma-paraganglioma (PPGL), and primary hyperaldosteronism are rare. PPGL is a rare catecholamine-secreting neuroendocrine tumour arising from the adrenal gland in 80–85% or extra-adrenal chromaffin cells of the autonomic neural ganglia in the remainder. The annual incidence of PPGL is 3–8 cases per million per year in the general population. Catecholamine-induced cardiomyopathy (CICMP) has got a prevalence of 8–11% among patients with PPGL. Hypertension, either sustained or episodic, is present in the vast majority (95%) of PPGL patients. However, among patients with CICMP, hypertension is present only in 65% of cases and the classical triad of paroxysmal headache, sweating, and palpitation is present only in 4%. Based on the cardiac remodelling in response to endogenous catecholamine excess, PPGL patients might present with one of the three CICMPs, including dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), or Takotsubo cardiomyopathy (TCM). Regardless of the subtypes, all CICMPs have many features in common — a dramatic clinical presentation, reversible cardiomyopathy, similar repolarisation electrocardiography changes, mild-moderate cardiac biomarker elevation, and normal coronary arteries on coronary angiography. CICMP should be suspected in patients with non-ischaemic, non-valvular forms of cardiomyopathy, even in those without definite features of catecholamine excess. PPGL associated TCM should be suspected in all acute coronary syndrome (ACS) patients exhibiting pronounced blood pressure variability with no culprit lesions on coronary angiography. This article will provide a review of the various CICMPs, their pathophysiology, clinical features, and the management options.
Cardiomyopathies are myocardial disorders characterized by structural/functional abnormalities of the cardiac muscles sufficient to cause observed myocardial abnormality in the absence of coronary artery disease (CAD), hypertension, valvular heart disease and congenital heart disease [1]. The cardiac muscle disorders caused by CAD, hypertension, valvular heart disease and congenital heart disease were considered as exclusion criteria because their diagnosis and management were quite distinct from most other cardiomyopathies. Nevertheless, ischaemic cardiomyopathy, hypertensive cardiomyopathy, and congenital heart disease with co-existent cardiomyopathy are well-known entities [2, 3, 4, 5]. Ischaemic cardiomyopathy is characterized by left ventricular systolic dysfunction in patients with previous myocardial infarction, myocardial revascularisation, or more than 75% stenosis in the left coronary artery main stem or left anterior descending artery [2]. Hypertensive cardiomyopathy is characterised by left ventricular hypertrophy associated with left ventricular diastolic and/or systolic dysfunction in hypertensive patients, in the absence of other conditions known to cause left ventricular hypertrophy or left ventricular dysfunction [3]. Congenital heart disease can co-exist with cardiomyopathy as the mutations that are known to cause congenital heart disease can also result in cardiomyopathy [4].
The European Society of Cardiology (ESC) working group in 2008 grouped cardiomyopathies into specific morphological and functional phenotypes including hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and unclassified cardiomyopathies, the latter including left ventricular non-compaction (LVNC) and Takotsubo cardiomyopathy (TCM) [1]. Each of these phenotypes can be subclassified into a genetic form (associated with single gene mutations) and a non-genetic form. The genetic form is mostly familial but can rarely be caused by de novo mutations. The non-genetic form can either be idiopathic (with no identifiable cause) or acquired (complication of other diseases) [1, 5]. A classification proposed by the World Heart Federation in 2013, known as MOGE(S) incorporates the morpho-functional phenotype (M), organ involvement (O), genetics (G), etiology (E) and the functional status (S) of the disease [6]. According to this classification, the term “idiopathic cardiomyopathy” is no-longer appropriate, as the cause and the mechanisms are increasingly identified in a significant proportion of cardiomyopathies [7].
Many endocrine diseases are associated with acquired cardiomyopathies and heart failure [8]. The phaeochromocytoma and paraganglioma (PPGL) are associated with DCM (38.7%), TCM (23.3%), inverted TCM (19.6%), HCM (6.1%), myocarditis (4.9%), and unspecified cardiomyopathy (8.6%) [9]. Severe hypothyroidism [10], thyrotoxicosis [11], primary hyperaldosteronism [12, 13], Cushing’s syndrome [14], acromegaly [15], hypocalcaemia [16] and vitamin D deficiency [17] are associated with DCM. Carcinoid syndrome [18] is associated with RCM, whereas lipodystrophy syndrome (both generalised and partial lipodystrophy) [19] is associated with predominantly HCM and rarely DCM.
PPGLs are rare catecholamine-secreting neuroendocrine tumours arising from either the adrenal gland or the extra-adrenal chromaffin cells of autonomic neural ganglia [20]. Catecholamine-induced cardiomyopathies (CICMPs) are rare, dreadful, and difficult-to-treat complications of PPGL with an unexpectedly high surgical risk [21]. CICMP has got a prevalence of 8–11% among patients with PPGL [22, 23]. This is a potentially reversible cause of cardiomyopathy. With an early diagnosis, adrenergic blockade, and timely surgical treatment the prognosis is quite favorable in most cases.
Catecholamines are tyrosine-derived neurotransmitters including dopamine, norepinephrine, and epinephrine [24, 25]. They also act as hormones regulating key physiological processes across nearly all body tissues, and thus are involved in the pathogenesis of many systemic diseases [24, 25, 26]. They are predominantly synthesised by the chromaffin cells of the adrenal medulla and sympathetic postganglionic neurons. The adrenal medulla releases epinephrine (80%) and norepinephrine (20%) in response to its sympathetic supply, commonly as part of an acute stress response. This release is in conjunction with norepinephrine secretion from sympathetic nerve terminals. The catecholamines have a short half-life of 1–3 minutes and are deaminated by monoamine oxidases or methylated by catechol-O-methyl transferases. These enzymes are also found in other tissues such as the liver [25].
The concentration and type of catecholamine determine the cardiovascular
response to it. Acutely, catecholamines increase heart rate, systemic vascular
resistance and myocardial contractility while reducing venous compliance [23].
They are considered the most potent positive inotropic agents in relation to the
heart and are the main regulators of cardiac output. Epinephrine also acts as a
bronchodilator and increases the rates of glycogenolysis, lipolysis and oxygen
consumption [25]. In the heart, the norepinephrine that is released upon
activation of sympathetic nervous system acts through
The
Catecholamine excess states have been implicated in the pathogenesis of various
types of cardiomyopathies including tachycardia-related cardiomyopathy, HCM, DCM,
and TCM. The catecholamine excess may also aggravate pre-existing cardiac
conditions in patients with phaeochromocytoma [28, 29, 30, 31]. Fig. 1 summarizes the
major pathophysiological mechanisms of catecholamine-induced cardiomyopathy.
Cardiomyocyte remodelling following sustained catecholamine excess leads to the
development of different types of CICMPs. Acute excessive adrenergic stimulation
may result in overstimulation of
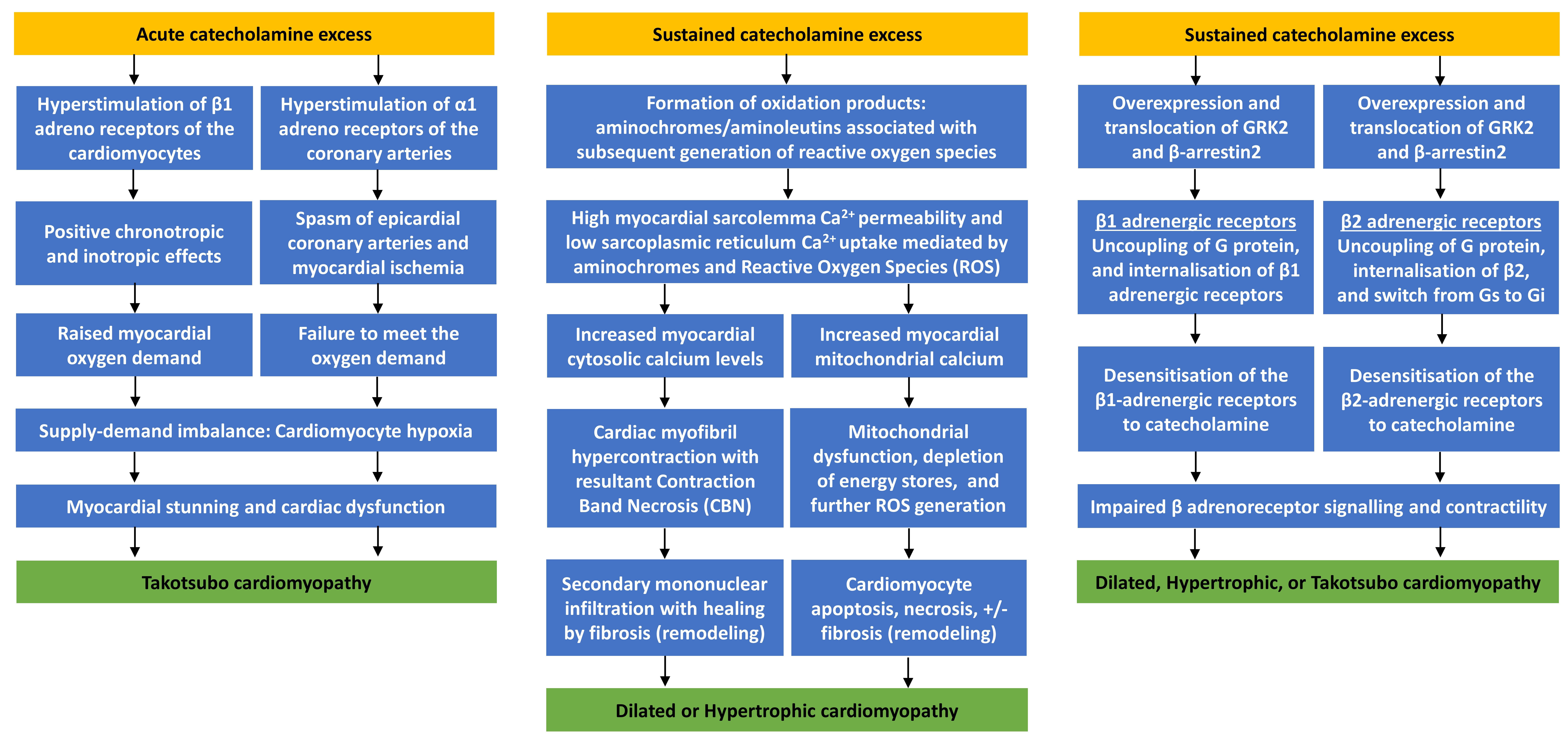 Fig. 1.
Fig. 1.Major pathophysiological mechanisms of catecholamine-induced
cardiomyopathy. Catecholamine excess states are associated with supply-demand
imbalance, myocardial stunning ad cardiac dysfunction. Catecholamine excess
states are associated with formation of oxidation products and reactive oxygen
species resulting in cardiomyocyte apoptosis, necrosis, and cardiac remodelling
by fibrosis. Finally, catecholamine excess states are also associated with
impaired
Prolonged exposure to raised catecholamine levels leads to blunting of the
 Fig. 2.
Fig. 2.Proposed mechanism for
The Gs to Gi switch is described only with
Catecholamines produce a direct toxic effect on the myocardium by increasing the sarcolemmal permeability and cellular calcium influx [28, 31]. These deleterious effects of catecholamine excess are mediated by the oxidation products of catecholamines rather than catecholamines per se [41]. Under physiological conditions catecholamines are metabolized mainly by catechol-O-methyl transferase and monoamine oxidase pathways. However, under conditions of catecholamine excess, these pathways are overwhelmed and the catecholamines (epinephrine released from adrenal medulla and norepinephrine released from sympathetic nerve endings) undergo oxidation to form aminochromes (active metabolite) and aminoleutins (inactive metabolite). The oxidation of catecholamines into aminochromes and aminoleutins are associated with the generation of reactive oxygen species (ROS) [41]. The oxidative stress resulting from the ROS molecules together with the aminochromes leads to calcium handling abnormalities inside the cardiomyocytes including high calcium permeability through the sarcolemma and low calcium uptake by the sarcoplasmic reticulum (SR) [42]. The resultant increase in myocardial cytosolic and mitochondrial calcium levels causes mitochondrial dysfunction, depletion of energy stores, and augments the ROS generation culminating in myocardial apoptosis, necrosis, with/without fibrosis. Similarly, the raised intracellular calcium levels mediated by the aminochromes inside the vascular smooth muscle cells results in coronary spasm, myocardial ischaemia, ventricular arrhythmias, and contractile dysfunction [43].
Catecholamine-mediated myocardial stunning has been implicated in the
pathogenesis of TCM. The name TCM came from the shape of cardiac apex that
resembles ‘takotsubo’, a conical Japanese pot used for catching octopus. Inverted
TCM with apical sparing has also been described [44, 45]. In the mammalian left
ventricle (LV), there is an apical-basal gradient for
In contrast to the above theory of epinephrine mediated Gs to Gi switch, a systematic review and meta-analysis of 156 published case reports of TCM patients where the TCM was induced by exogenous epinephrine, exogenous norepinephrine, or endogenous catecholamine (PPGL) did not find any direct causal relation between epinephrine and TCM [47]. However, epinephrine excess was found to have an indirect effect via hyperactivation of the sympathetic nervous system (SNS) which in turn leads to disruption in cardiac sympathetic nerve terminals with norepinephrine spillover [48, 49]. This would explain the multifocal histological changes (contraction band necrosis) and patchy cardiac MRI changes seen in PPGL-related CICMPs [50]. Moreover, the SNS hyperactivation can explain the LV basal hypokinesia seen in inverted TCM.
Excess catecholamines that are released directly into the myocardium via sympathetic nerve terminals are more toxic in comparison to the excess catecholamines reaching the myocardium via blood stream [50]. Increased cardiac SNS activity, increased norepinephrine synthesis/release, decreased norepinephrine reuptake by the terminal nerve axons, increased sensitivity to catecholamines, myocardial ischaemia due to epicardial coronary artery spasm, and transient LV outflow tract obstruction have all been suggested as possible pathophysiologic mechanisms for TCM [51, 52, 53, 54]. Additionally, oestrogen-induced downregulation of the adrenergic receptors of the heart may also have a role, as TCM is more common in postmenopausal women [53, 55].
PPGLs are rare catecholamine-secreting neuroendocrine tumours arising from either the adrenal gland or the extra-adrenal chromaffin cells of autonomic neural ganglia. The annual incidence of phaeochromocytoma has been estimated as 3–8 cases per million per year in general population, and it occurs in less than 0.5% of patients with hypertension [56, 57]. The frequency can be higher in those with adrenal incidentalomas, where up to 4.2% can have PPGL [58]. Phaeochromocytomas can be sporadic or familial. Sporadic forms usually present between the ages of 40–50 years, whereas the hereditary forms including multiple endocrine neoplasia type 2 (MEN2), neurofibromatosis type 1 (NF1), von Hippel-Lindau syndrome (VHL), and succinate dehydrogenase (SDH) subunit B, C and D mutations (SDHB, SDHC, and SDHD, respectively), usually present in childhood or early adulthood [59]. The hereditary PPGLs related to VHL mutations secrete norepinephrine, SDH mutations secrete dopamine and norepinephrine, whereas RET mutations (MEN2A) secrete norepinephrine and epinephrine [60].
These tumours secrete catecholamines including epinephrine, norepinephrine, and dopamine. Phaeochromocytomas arising from the adrenal medulla accounts for nearly 80–85%, whereas the paragangliomas arising from autonomic neural ganglia account for 15–20% of PPGL [60]. While normal adrenals predominantly secrete epinephrine, majority of PPGLs predominantly secrete norepinephrine with only about 15% secreting predominantly epinephrine [61]. The PPGLs predominantly secreting dopamine are rare and these are malignant PPGLs in which the dopamine decarboxylase enzyme is missing [62]. Usual presenting features of PPGL include the classical triad (headache, hyperhidrosis, and palpitation), tachycardia, tremor, anxiety, pallor, chest pain, dyspnea, and sometimes local tumoral symptoms like abdominal pain, although many patients can be asymptomatic. Hypertension is present in nearly 95% of patients with PPGL, with sustained hypertension noted in 55% and episodic hypertension in 45% [63].
The PPGLs predominantly secreting norepinephrine presents with sustained hypertension, those predominantly secreting epinephrine presents with episodic hypertension, and those predominantly secreting dopamine presents with no hypertension [61]. Fluctuating hypertension with cyclic bouts of hypertension and hypotension is well described in phaeochromocytoma patients possibly due to spontaneous infarction of the tumour during the phaeochromocytoma crisis [64, 65, 66, 67]. The proposed mechanisms for spontaneous necrosis include catecholamine-induced vasoconstriction, embolic infarction, haemorrhage into the tumour, high intracapsular pressure, or antihypertensive therapy induced hypotension [67]. Hypertension can be absent in PPGL patients, and the various proposed mechanisms include secretion of dopamine as the predominant catecholamine, conversion of catecholamines into inactive metabolites within the tumour itself, secretion of other molecules together with catecholamines - including vascular endothelial growth factor that suppresses catecholamine action, reduction in sensitivity of receptors to catecholamine, or significant reduction in cardiac function due to CICMP [68].
Cardiovascular complications of phaeochromocytomas include left ventricular
hypertrophy, ACS including myocardial infarction, acute pulmonary oedema,
myocarditis, cardiac arrythmias, cardiomyopathies, thromboembolism, and shock
[21, 57, 69]. Based on the cardiac remodelling in response to the endogenous
catecholamine excess, PPGL patients might present with either one of the three
types of CICMPs, including DCM, HCM, or TCM [70]. DCM characterised by congestive
cardiac failure, pulmonary oedema or cardiogenic shock associated with diffuse
left ventricular or even biventricular dysfunction can be one of the presenting
manifestations of PPGL [71]. DCM can occur spontaneously or can occur after
initiation of
TCM patients often present with acute chest pain, acute severe heart failure, dynamic ST-T changes, elevated cardiac biomarkers, and left ventricular regional wall motion abnormalities (RWMA) in a circumferential apical, midventricular, or basal distribution, without evidence of significant CAD on coronary angiographic studies [77]. Thus, TCM closely mimics ACS and hence these patients are often misdiagnosed and treated as ACS. Nearly 1–3% of all patients presenting with suspected ST-segment elevation myocardial infarction has in fact TCM [77]. The diagnostic hallmark of TCM is the reversibility of cardio depression occurring within weeks of initial presentation [57].
The clinical picture of TCM associated with PPGL and that not associated with PPGL are quite divergent. Nearly two-thirds of TCM patients associated with PPGL develop cardiac complications in comparison to only one-fifth of TCM patients without PPGL. The TCM-PPGL patients are associated with higher recurrence rate (17.7% vs 3.26%). These patients are younger (fourth or fifth decade). The TCM localisation pattern was different in TCM-PPGL patients, with the basal pattern contributed to almost 1/3 of cases and global pattern contributed to 1/5 of the cases [78]. The contributions of TCM-PPGL to apical, mid-ventricular, basal, and global TCM patterns were 43.9%, 5.6%, 26.2%, and 20.6%, whereas the corresponding contributions of TCM cases in general were 81.7%, 14.6%, 2.2%, and 0% respectively [47]. When compared to the classical TCM pattern, the inverted TCM that was commonly seen in TCM-PPGL was associated with higher complication rates, including, cardiogenic shock, heart failure, acute renal failure, and arrhythmias. This higher complication rates could be related to the higher levels of circulating catecholamines in TCM-PPGL patients. Moreover, the precipitating factors are often not clear in TCM-PPGL patients [79].
A catecholamine crisis or phaeochromocytoma multisystem crisis is a rare, acute, severe complication of catecholamine-induced haemodynamic compromise and collapse [80]. The term ‘type A crisis’ is used to describe a more limited crisis without sustained hypotension, whereas ‘type B crisis’ is used to describe a severe presentation with sustained hypotension, shock and multi-organ dysfunction [45]. Fever can be present in a minority of cases without a focus of sepsis, possibly related to secretion of interleukin-6 by the tumour [81, 82, 83]. Phaeochromocytoma crisis can mimic other conditions leading to misdiagnosis and phaeochromocytomas are often found as an incidental finding on imaging performed for an alternative diagnosis [44, 84]. Nearly 11% of phaeochromocytoma patients required in-hospital treatment on intensive care units due to complications caused by unsuspected phaeochromocytomas [85]. Phaeochromocytoma-crisis is associated with an overall mortality of 15%, with a higher (28%) mortality in those developing ‘type B crisis’ compared to those developing ‘type A crisis’ (6%) [45].
The diagnosis of CICMP is often delayed due to atypical presentations. In a study on 163 PPGL patients with CICMP, hypertension was the presenting symptom only in 65% of patients [86]. Among the various CICMP subgroups, hypertension was common with DCM (83%), and HCM (80%), compared to TCM. The classical PPGL triad of headache, sweating, and palpitation was present in only 4% of patients. Among the subgroups, the classical triad was common in HCM patients, whereas the DCM patients commonly presented with congestive heart failure [86]. Hence, CICMP should be suspected in all cardiomyopathy patients that is non-ischaemic, and non-valvular, even in those without definite features of catecholamine excess. Similarly, PPGL associated TCM should be suspected in all ACS patients exhibiting pronounced blood pressure variability with no culprit lesions on coronary angiography. The diagnosis is important as surgical removal of PPGL would improve CICMP in 96% of cases. Moreover, if not undergoing surgical resection, the disease would lead to mortality or cardiac transplantation in 44% of cases [86].
Consider possibility of catecholamine-induced cardiomyopathy when a known PPGL
patient presents with symptoms of heart failure, hypotension, and multisystem
crisis [21]. However, the diagnosis of CICMP can often be missed initially in
patients who are not already known to have a phaeochromocytoma [87]. Fig. 3 (Ref.
[60]) shows an algorithm for the diagnostic workup of PPGL. In asymptomatic
patients, the phaeochromocytoma often comes to light whilst investigating for an
adrenal incidentaloma with non-contrast computed tomography (CT) with an
attenuation value of more than 10 Hounsfield units (
 Fig. 3.
Fig. 3.An algorithm for the diagnostic workup of phaeochromocytoma and
paraganglioma [60]. This algorithm should be used in the evaluation of people
who are either symptomatic for PPGL or who are asymptomatic, but with presence of
adrenal incidentaloma, hypertension in young non-obese people, or hereditary
predisposition for PPGL. Abbreviations: PPGL, Phaeochromocytoma and
Paraganglioma; ULN, Upper Limit of Normal; CT, Computed Tomography;
The 2016 European Endocrine Society guidelines recommend the addition of 3-methoxytyramine (the dopamine metabolite) in the initial screening panel for the diagnosis and follow-up of patients with PPGLs [89]. The addition of 3-methoxytyramine along with free metanephrines would improve the sensitivity (97.2% vs 98.6%) but reduce the specificity (95.9% vs 95.1%) [90]. The dopamine producing malignant PPGLs are not missed with this addition. The 2016 European Endocrine Society guidelines also recommend using plasma chromogranin as a screening tool in cases with a clinical probability for PPGL once plasma free metanephrines and 3-methoxytyramine are negative [89]. Plasma and urinary catecholamines and urine vanillylmandelic acid (VMA) are less often used currently because of suboptimal sensitivities and specificities [60].
Cardiac failure due to any cause would be associated with catecholamine release, with a likely consequent rise in plasma metanephrine levels. As metanephrine levels are not yet validated in patients with cardiac failure, routine use of serum fractionated metanephrines or urinary free metanephrines in all cardiomyopathy patients would result in significantly high false-positive results [91]. These tests should be used only when the pre-test probability of CICMP is high. The factors that increase the pre-test probability of CICMP include younger age at diagnosis of cardiomyopathy; genetic diseases associated with PPGL (MEN2, NF1, VHL, SDH); patients with symptoms suggestive of PPGL including classical PPGL triad; blood pressure (sustained, episodic, or fluctuating blood pressure); inverted TCM (with higher risk for cardiogenic shock, heart failure, acute renal failure, and arrhythmias); and adrenal incidentalomas with radiological features of phaeochromocytoma [91].
These patients with clinical features suggestive of PPGL or CICMP should undergo anatomical localisation studies using computed tomography (CT) or magnetic resonance imaging (MRI) after the initial step of biochemical confirmation. CT should be the first-choice imaging modality because of its excellent spatial resolution for thorax, abdomen, and pelvis. However, MRI is recommended in patients with residual/recurrent/metastatic PPGL, for detection of skull base and neck paragangliomas, in patients with surgical clips that could cause artifacts when using CT, in patients with an allergy to CT contrast, and in patients in whom radiation exposure should be limited (children, pregnant women, those with known germline mutations, and those with recent excessive radiation exposure) [87].
The anatomical imaging should be followed by functional imaging. The
Iodine-123-tagged MIBG (metaiodobenzylguanidine) scintigraphy can detect tumours
that were undetected by CT or MRI and is useful to establish the diagnosis of
PPGL in cases where tumours were already detected on anatomical imaging [92]. The
Electrocardiogram (ECG) and 2-dimensional echocardiogram should be performed in all cases of suspected or confirmed PPGL/CICMP. Electrocardiographic changes range from unremarkable, sinus tachycardia, wandering pacemaker, and repolarisation ECG changes. The latter include ST-segment elevation, ST-segment depression, peaked T-waves, or giant inverted T-waves. The ST-segment elevation with giant inverted T-waves is seen in apical TCM, whereas ST-segment depression with peaked T-waves is seen in basal TCM [69]. The echocardiographic features of different CICMPs are given in Table 1 [21, 94]. In TCM, echocardiography helps to identify the pattern and extent of disease. In other CICMPs, echocardiography helps to assess mechanical complications including dynamic LV outflow tract obstruction, with or without systolic anterior motion of the anterior mitral leaflets, and/or functional mitral valve regurgitation.
| Type | Echocardiographic features various catecholamine-induced cardiomyopathies |
| DCM | Dilated LV and possibly RV; no or minimal eccentric left ventricular hypertrophy with or without LVSD; global hypokinesia is common, but RWMA can occur |
| HCM | Concentric hypertrophy; normal or reduced internal chamber dimensions with or without systolic anterior motion of anterior mitral valve leaflet (SAM) |
| TCM | Classic TCM: apical ballooning (apical akinetic segment, hyperkinetic base) |
| Inverted TCM: akinetic base/mid-ventricular segment, hyperkinetic apex | |
| Mid-ventricular TCM: normal apex and base, mid-ventricular dyskinesia | |
| Localized TCM: segmental dyskinesia in coronary artery distribution |
Coronary angiogram and cardiac MRI should be performed in selected cases with cardiovascular involvement [87]. Left ventriculography performed during coronary angiogram should not be considered as the sole investigation to assess the myocardial function, as right ventricle (RV) could be involved in one-fourth of the TCM cases [95]. Cardiac Magnetic Resonance (CMR) can differentiate TCM from myocarditis and ischaemic cardiomyopathy. CMR in patients with TCM exhibits high-intensity signal on T2-weighted images in a transmural distribution, without significant late gadolinium enhancement, in regions with wall motion abnormalities. Occasionally, the CMR in TCM patients might show areas of myocarditis, focal fibrosis as small foci of late gadolinium enhancement [96].
PPGL was one of the exclusion criteria for the diagnosis of TCM as per the Mayo
Clinic criteria that has been used historically [97]. However, in 2018 the Heart
Failure Association-European Society of Cardiology (HFA-ESC) TCM task force
developed an international takotsubo diagnostic criteria, which is also known as
InterTAK Diagnostic Criteria, wherein PPGL was included as the secondary cause of
TCM [77]. An InterTAK diagnostic score can be calculated as given in Table 2.
Patients with symptoms suggestive for an ACS, however, with no ST-elevations in
the ECG, but with high probability of TCM should undergo screening using the
InterTAK Diagnostic Score. If the score is positive with
| International Takotsubo diagnostic criteria | InterTAK diagnostic score |
| Female sex | 25 points |
| Emotional stress | 24 points |
| Physical stress | 13 points |
| No ST-segment depression | 12 points |
| Psychiatric disorders | 11 points |
| Neurological disorders | 9 points |
| QTc-Interval prolongation | 6 points |

High index of suspicion and prompt diagnosis is important for a favourable
outcome from this potentially fatal condition. The diagnosis should be considered
in all patients presenting with acute cardiomyopathy and cardiogenic shock
without a clear ischaemic or valvular aetiology [99]. The diagnosis should be
confirmed immediately as several medications including dopamine D2 receptor
antagonists,
The initial management of phaeochromocytoma crisis should be in a setting where
expertise and access to mechanical circulatory support is available.
Stabilisation of blood pressure with
The
 Fig. 5.
Fig. 5.An algorithm for the management of phaeochromocytoma and
paraganglioma [60]. Laparoscopic adrenalectomy, either conventional or
robot-assisted, is the gold standard technique for adrenalectomy in tumors
In patients with acute decompensated cardiomyopathy, phentolamine given as
intravenous bolus or infusion is preferred because of its short half-life and
rapid reversal of adverse effects. Phenoxybenzamine, which has a half-life of 24
hours, can be started once haemodynamic stability has been achieved [94].
Doxazosin is a competitive
Management of patients presenting with acute heart failure requires adequate diuretics, but this is often complicated by hypotension due to intravascular volume depletion or systolic heart failure [21]. Indiscriminate use of inotropes and vasopressors should be avoided as they precipitate a phaeochromocytoma-crisis and may cause cardiogenic shock by causing LV outflow tract obstruction. Hence these drugs should be initiated judiciously, utilizing the lowest possible dose in patients with CICMP and hypotension. Fluid status needs to be monitored carefully and intra-arterial balloon counter-pulsation, the Impella system, the TandemHeart device or veno-arterial extra-corporeal membrane oxygenation (VA ECMO) can be considered in refractory cases of catecholamine crisis associated with hypotension [44, 104, 105, 106, 107, 108]. Severe hypotension may occur once the tumour is resected, which might result from the abrupt cessation of catecholamine secretion and depleted blood volume.
Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers may have a role in managing heart failure in patients with phaeochromocytoma. Clinical signs of congestive heart failure may resolve within two weeks following treatment with captopril. Myocardial performance can also become normal within two weeks of medical therapy, with one week with captopril monotherapy and with another week with captopril-phenoxybenzamine combination therapy [109]. Proposed mechanisms include the inhibition of the cardiac renin-angiotensin axis and inhibition of free radicals from long chain fatty acids described in phaeochromocytoma [110, 111]. Orthostatic hypotension in combination with sustained hypertension has been reported to be a feature in 10–50% of the individuals with phaeochromocytoma [112]. Treatment should primarily focus on restoring circulating volume. Mineralocorticoids should not be administered in patients with phaeochromocytoma [100, 101].
In summary, there is no specific treatment approach for the management of
patients with CICMP. The preoperative management of CICMP should include control
of blood pressure variability by lowering sympathetic activation with the help of
Studies have shown that sustained catecholamine excess is associated with cardiac remodelling characterized by cardiomyocyte hypertrophy, apoptosis, and fibrosis mediated by coronary ischaemia, increased mechanical stress, cytokines and neurohormones [115]. Catecholamine excess is associated with inflammation and aging (senescence) of cardiomyocytes, upregulation of tumour suppressors including p53, and generation of adhesion molecules, all of which together mediating the cardiac dysfunction [116]. Animal studies have also shown that catecholamine-induced lipotoxicity is one of the mechanisms for causing myocardial dysfunction and TCM [117].
In response to endogenous catecholamine excess, PPGL patients might present with
one of the three CICMPs, including DCM, HCM, or TCM. Regardless of the subtypes,
all CICMPs have many features in common — a dramatic clinical feature, reversible
cardiomyopathy (if diagnosed early, optimally treated: medically or surgically),
similar repolarisation electrocardiographic changes, mild to moderate cardiac
biomarker elevation, normal coronary angiography, and focal or multifocal
contraction band necrosis with secondary mononuclear cell infiltrations on
histology. CICMP should be suspected in all non-ischaemic, and non-valvular
cardiomyopathy patients, even in those without definite features of catecholamine
excess. Similarly, PPGL-associated TCM should be suspected in all ACS patients
exhibiting pronounced blood pressure variability with no culprit lesions on
coronary angiography. There is no specific treatment approach for the management
of CICMP. The preoperative management of CICMP should include control of blood
pressure variability by lowering sympathetic activation with the help of
• Suspect CICMP in patients with non-ischaemic and non-valvular acute
cardiomyopathy, or in patients with inverted TCM, especially in those with young
age at diagnosis, genetic diseases, marked blood pressure variability, with or
without PPGL related symptoms. • Cardiac failure due to any cause would result in catecholamine release, with a
consequent falsely elevated plasma fractionated metanephrine levels. Hence these
tests should be used only when the pre-test probability of CICMP is high. • With an early diagnosis, adrenergic blockade, and timely surgical treatment
using the optimal anaesthetic techniques, the prognosis is quite favorable most
of the time.
AK drafted the paper initially with advice & Guidance from JMP and CJF who further developed, edited and approved it in the current form with mutual agreement between all the authors.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.