


 , Gisli Jenkins 1,*
, Gisli Jenkins 1,*
1 Margaret Turner Warwick Centre for Fibrosing Lung Disease, National Heart and Lung Institute, Imperial College London, SW3 6LY London, UK
Abstract
Idiopathic pulmonary fibrosis (IPF) is the prototypical fibrosing interstitial lung disease, characterised by its unrelenting progressive course and poor prognosis. The incidence of IPF is rising and is becoming a major public health concern. Debilitating dyspnoea and respiratory failure results. Death occurs on average 3–5 years from the time of diagnosis. Clearer understanding of the pathobiology of the condition continues to advance and although we may now better understand disease mechanisms, our therapeutic approach remains limited. In the UK, only two treatments are licensed for IPF, and both can only slow the process down. An ideal silver bullet would halt and ideally reverse established fibrosis. New therapies are showing promise; however, lung transplant remains the only treatment that can substantially increase both duration and quality of life. Here we will provide a comprehensive overview of IPF to summarise definitions, epidemiology, mechanisms, diagnostics, and management.
Keywords
- idiopathic pulmonary fibrosis
- risk factors
- diagnosis
- outcomes
- management
Idiopathic pulmonary fibrosis (IPF) is a fibrotic interstitial lung disease (fILD) which is characterised by scarring of the lung parenchyma. Not all interstitial lung diseases (ILDs) result in scarring, for example, acute hypersensitivity pneumonitis, which may resolve when the inciting antigen is removed. If an ILD is prone to develop scarring, it is referred to as an fILD. Some may be associated with a clear aetiology, such as asbestosis or a connective tissue disease (CTD) or an assumed aetiology as observed in fibrotic hypersensitivity pneumonitis (fHP). However, if there is a characteristic clinical, radiological and pathological pattern of parenchymal fibrosis in the absence of an identifiable aetiological factor, it is termed IPF [1].
IPF is defined by the finding of usual interstitial pneumonia (UIP) either histologically, or, more commonly, by a characteristic radiological pattern, which obviates the need for a biopsy to reach a diagnosis. Histological UIP is characterised by the presence of fibroblastic foci and is also associated with honeycomb cysts, but is not specific to IPF; therefore, the clinical context described above is crucial for a diagnosis of IPF [2].
A confident diagnosis of IPF is paramount not only for management considerations but also because the disease follows a typical progressive course of accelerated and irreversible lung damage with loss of lung function leading to respiratory failure and death. This therefore has huge prognostic implications for patients. In addition, treatments used in other forms of idiopathic interstitial pneumonias (IIP) are frequently harmful in IPF [3].
Non-respiratory clinicians should have an awareness of IPF for the following reasons: the incidence of IPF is rising; there is a huge burden of asymptomatic parenchymal fibrosis picked up incidentally (for example, during lung cancer screening programmes), some of which will progress to IPF, where the morbidity and mortality are unacceptably high; and IPF can be associated with multiorgan fibrosis, including bone marrow and liver, which can be seen in short telomere syndromes [4].
In the subsequent sections, the epidemiology, risk factors, diagnostics, and management considerations of IPF will be described.
Prevalence estimates are challenging in IPF in part due to global differences in diagnostic approaches, in addition to underestimation, as cases are often diagnosed in advanced states of disease. Males are affected more than females [5]. Global prevalence is currently estimated between 0.33 and 4.51 per 10,000 persons worldwide [6]. This vast range can be explained in part by studies from Greece and Taiwan, where prevalence is very low, and South Korea, where it is amongst the highest in the world [7]. Whether these estimates are reflective of true differences in prevalence is uncertain, as there are clear discrepancies between these regions in average age of the population (high in South Korea with a male sex preponderance), estimated mortality (very high in Taiwan which may reflect late diagnosis from lack of disease awareness), and a difference in approach to IPF diagnosis. When these studies are excluded, estimates in the UK are in the region of 0.78 per 10,000 persons and the USA 2.4 per 10,000 (although the range here is 0.67–11.1). Differences here are likely to be accounted for by heterogeneity in study design and differences in included patient groups. These disparities aside, it is estimated that 8000–9000 new cases of IPF are diagnosed in the UK each year [8]. The incidence is rising in the UK [9].
IPF is a disease of aging with increasing incidence in older people. The median age of diagnosis is 65 years in interventional clinical trials and 72 years of age in registries and observational trials [10]. Furthermore, cellular, molecular and genetic pathways associated with aging are found in IPF.
Survival remains poor. A retrospective analysis of U.S. patients reported a median survival of 2.8 years from diagnosis [11] which is supported by UK cohort study of approximately 1500 patients with median survival of 2.7 years [9]. However, survival estimates do vary, with a median survival of 4.5 years reported in a Finnish registry which was attributed to only mild impairments in lung function in this cohort [12]. Multiple variables may be associated with worse outcomes, including: increasing age, poor lung function and radiological extent of disease [13]. Optimistically, antifibrotic use does appear to improve survival outcomes and may reduce the risk of death [14]. However, no large-scale randomised controlled trials (RCTs) have been powered to demonstrate this, and observational studies demonstrating survival may exaggerate such benefits through immortal time bias [15].
The alignment of genetic abnormalities, environmental triggers, and certain comorbid conditions in an individual will substantially heighten their risk of developing IPF. Such interactions are depicted in Fig. 1.
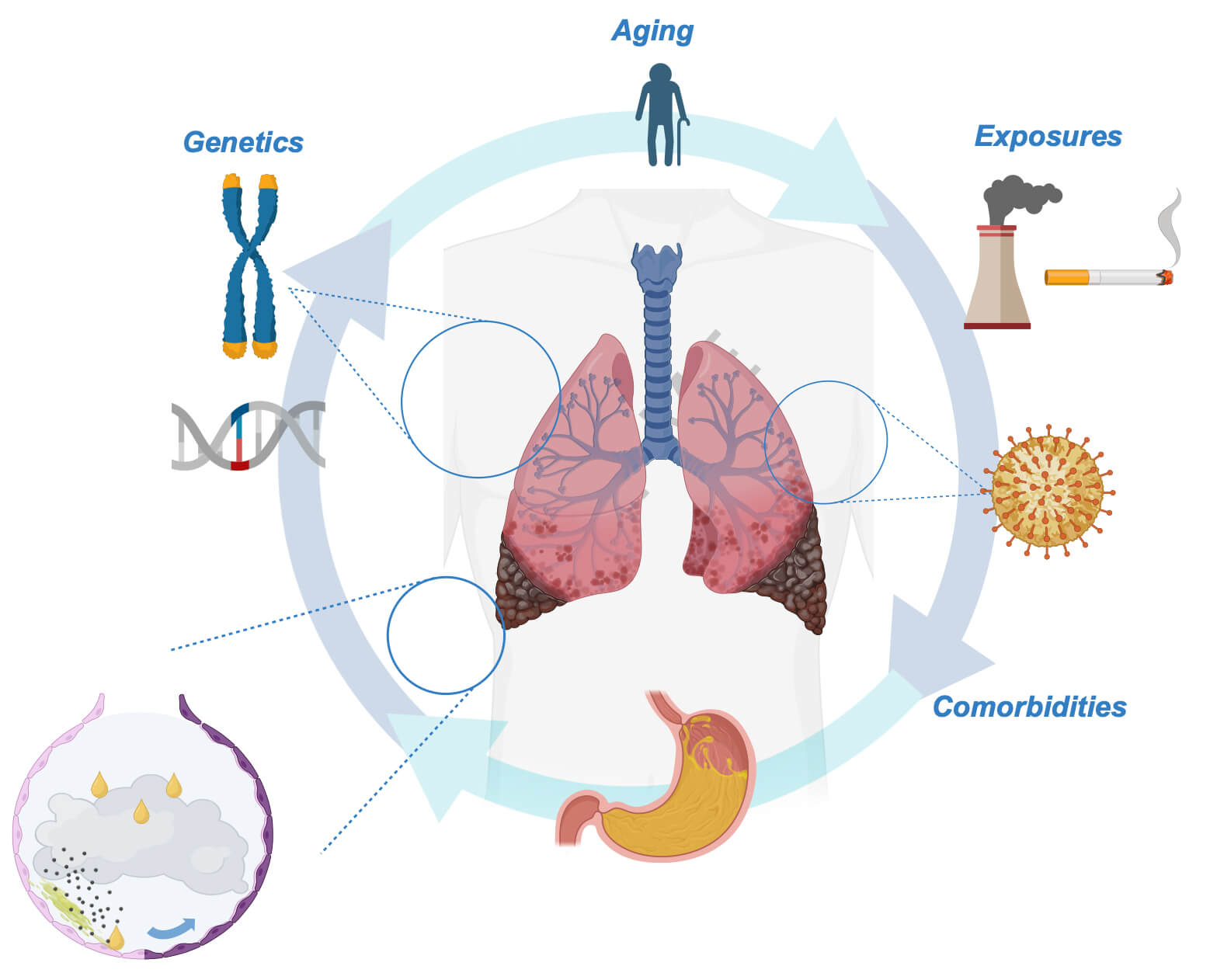 Fig. 1.
Fig. 1.
Patient, genetic, and environment factor interactions which increase IPF risk. The interplay between patient-specific factors, including comorbidities (including gastro-oesophageal reflux disease and chronic viral infection), environmental exposures (cigarette smoke, dust, etc.), and genetic abnormalities (MUC5B polymorphism, telomerase gene mutations, etc.), enhances an individual’s risk of developing IPF. Each risk factor in isolation is unlikely to drive the development of IPF. At the level of the alveolus, epithelial cells are “primed” by such genetic alterations and following a second hit from gastric contents or environmental pollutants, may initiate an exaggerated injury response with the evolution of pathological cell populations. The figure was drawn using BioRender.com. IPF, idiopathic pulmonary fibrosis; MUC5B, mucin 5B.
Genetic risk factors increase the risk of sporadic IPF caused by common allelic
variants (occurring in
In some cases, rare variants, defined by an allele frequency of
Patients suspected of having FPF should be offered genetic testing to guide screening and management in relatives if appropriate, especially given that the disease can follow a more aggressive course in successive generations. Table 1 lists the indications for genetic testing in the UK [19, 20].
| Indications for genetic testing |
| Newly diagnosed fILD with no cause or association in any patients |
| Positive family history in first or second degree relatives |
| Suspicion of underlying telomeropathy |
| • Unexplained blood dyscrasia (macrocytosis, anaemia, thrombocytopenia, leukopenia) +/- premature greying (significant hair greying |
| • Unexplained liver function disorder |
| • If being considered for lung transplant (as a tailored post-transplant immunosuppressive regimen may be required) |
fILD, fibrotic interstitial lung disease.
Environmental exposures through occupation or personal behaviours are crucial drivers of IPF, as recently reviewed by Johannson et al. [21], and are thought to increase the risk of developing IPF by 80% [22]. These include cigarette smoke exposure, occupational dusts, and pollution. A notable emerging risk is exposure to silica [23], and this may be increasingly seen amongst individuals who cut quartz kitchen worktops [24]. Finally, viral drivers have also been described. Chronic infection with the human herpesviruses can significantly elevate the risk of developing IPF [25]. Mechanisms described include the induction of profibrotic cytokine release and cellular changes which promote fibrosis, such as epithelial-to-mesenchymal transition (EMT), by chronic viral infection [26].
The risk of IPF may be further increased by the presence of other patient-specific factors, importantly, their comorbidities.
Gastro-oesophageal reflux has been reported to increase the risk of developing IPF by 60% and is on the causal pathway [27]. The effect of direct acid injury from gastric contents on the alveolus is a well described IPF driver (see Pathobiology section below). Symptoms of dyspepsia present in less than 60% of patients [28], and thus, reflux is “silent” in a large proportion. There is limited evidence to support the role of antiacid therapy on IPF-related mortality in symptomatic patients [29]. Surgical management of reflux in IPF patients is safe and well tolerated; however, it did not have a significant impact on lung function trajectory in a small randomised trial [30].
Diabetes is frequently seen in patients with IPF. One meta-analysis cites a frequency of 10–60% (the range attributed to included study design heterogeneity) [31]. The risk of developing IPF in diabetics is reported to be six-fold higher than in non-diabetics [32]. However, the relationship between diabetes and IPF is unlikely to be causal but instead associative, reflecting shared common risk factors for the development of both conditions [33]. Poor glycaemic control was associated with poorer forced vital capacity (FVC) [34], although the use of metformin in large-scale studies did not affect outcomes in IPF [35].
IPF is a risk factor for the development of numerous conditions, including cardiovascular disease, lung cancer, and chronic obstructive pulmonary disease (COPD). The risk of developing lung cancer is seven-fold higher in IPF than in matched controls [36]. Treatment is challenging as there is increased risk of acute exacerbation with treatment (surgery or chemo/radiotherapy) [37]. Similarly, the presence of COPD is common and results in higher mortality. This may be in part due to the increased risk of developing pulmonary hypertension in this subset of patients [38].
Pulmonary hypertension (PH) is common and associated with increased morbidity
and mortality in IPF. PH may be found in as many as half of patients with more
severe forms of disease [39]. IPF subjects with PH are
likely to have worse exercise tolerance than those without. However, the presence
of PH does not necessarily correspond with the severity of lung disease and
therefore is unlikely to be the result of hypoxic vasoconstriction alone. The
presence of PH results in poorer overall survival and may be as low as 10 months
when pulmonary artery pressure is
Systemic hypertension is the most common comorbidity in IPF. Prior corticosteroid use may be a risk factor; however, suggestion of a common vasculopathy has been reported [42]. Coronary artery disease may affect as many as 65% of patients and is associated with higher mortality [43].
A recent review by our group [44] provides greater mechanistic detail into how specific comorbidities may promote the development of IPF.
The current pathogenic paradigm is of a patient aging prematurely with the genetic risk factors suffering repeated microinjury (from cigarette smoke, dust, and acid) to the delicate alveolar epithelium which is lined by alveolar type I (AT1) cells. Abnormal airway mucin may be present, especially in those with polymorphisms in the MUC5B gene, “trapping” these inhaled toxins, thus sustaining the damage [45].
Under normal circumstances, alveolar type II (AT2) cells transdifferentiate to
replace damaged AT1 cells. In those with shortened telomeres or surfactant
protein gene disorders, what results is senescence and loss of normal AT2 cells
[46]. Senescent AT2 cells secrete the senescence-associated
secretory phenotype (SASP) proteins which include numerous proinflammatory and
profibrotic cytokines, including interleukin-1 beta (IL-1
In addition, AT2 cell loss leads to further abnormal populations of aberrant ‘fibrotic’ (in particular, basal) epithelial cells which line fibrotic regions of lung and communicate with mesenchymal cell populations [48]. Aberrant cell populations may arise not only from AT2 cells but also from small airways [49].
Abnormal epithelial cells begin to transition into mesenchymal cell populations with properties identical to fibroblasts in a process known as epithelial-to-mesenchymal transition (EMT) [50]. The key abnormal effector cell—the myofibroblast—may also be recruited from the circulation or activated from its resident position in the airway or interstitium. These cells are critical in normal wound repair processes but behave abnormally in IPF where they coalesce to form dense foci of cell populations, so-called fibroblastic foci and may secrete vast quantities of extracellular matrix (ECM): collagen I, III, and VI, as well as fibronectin.
As ECM is deposited, the density and stiffness of the tissue increase, changing
the lung’s biomechanical properties. This can lead to further tissue damage
through tissue stretch with each breathing cycle. Stretch of stiff tissue can
activate mechanotransduction pathways, including the liberation of transforming
growth factor beta (TGF
Vascular abnormalities and “fibrotic endothelial cells” are also described in IPF models and may have similar disease-driving features [44]. Dysregulated immune responses, including the emergence of populations of fibrotic alveolar macrophages, are also implicated and are capable of secreting proinflammatory and profibrotic cytokines [52].
As the fibrosis evolves and the lung stiffness increases, there is further distortion of lung biomechanical properties with increased ECM deposition and further parenchymal obliteration. These features combined lead to tissue distortion and destruction and show up as honeycomb cysts on computed tomography (CT) imaging.
The process is summarised in Fig. 2.
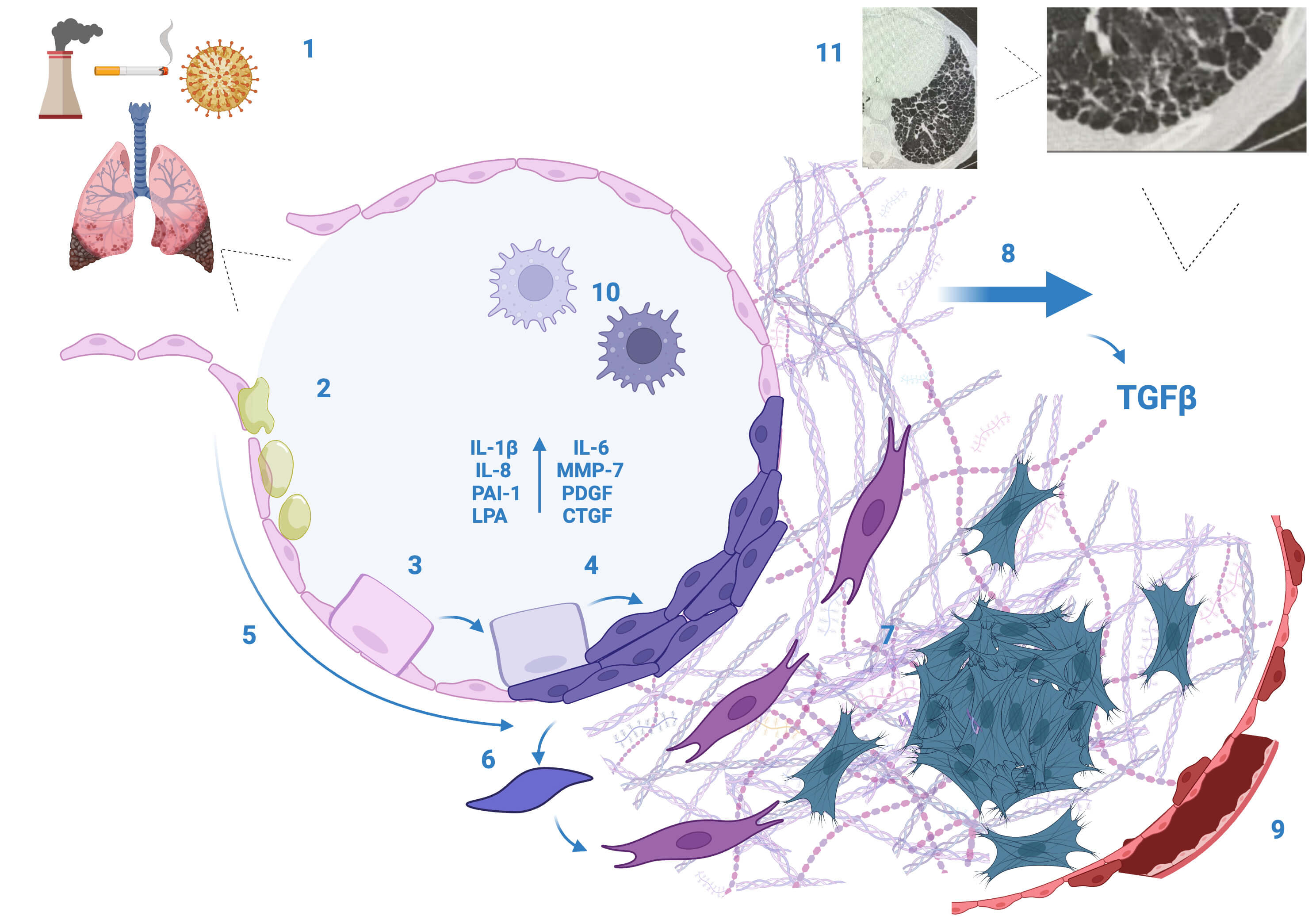 Fig. 2.
Fig. 2.
Schematic summarising the pathobiology of idiopathic pulmonary
fibrosis. (1) Known exposures, including cigarette smoke and dust, are inhaled
into the delicate alveoli. (2) Abnormal mucus (especially in those with MUC5B
polymorphisms) results in abnormal host-defence response. (3) Genetic
abnormalities can drive AT2 cell loss and/or senescence. (4) Aberrant fibrotic
epithelial cell populations may arise and line the alveoli from AT2 and (5) small
airways and (6) participate in EMT. (7) Fibroblasts and myofibroblasts (from EMT,
circulating cells, or resident cells) form dense focal cell populations and
secrete ECM. (8) With increasing ECM and stiffness, mechanotransduction pathways
are activated with a resultant increase in TGF
Patients are likely to report symptoms to their primary care practitioner in the years prior to diagnosis of IPF [53]. These symptoms are frequently attributed to old age, COPD, or heart failure. However, a lack of response to treatment and progressive worsening will prompt further investigation and ultimately a diagnosis of IPF.
The commonest symptoms reported are: fatigue (95%), dyspnoea (88%), and cough (85%), followed by weight loss [54]. These symptoms usually develop insidiously over time.
A proportion of patients will present in advanced states of disease and may be hypoxic at presentation (either on exertion or occasionally at rest). Finger clubbing is seen in up to 60% of cases and may be seen prior to the onset of respiratory symptoms [55]. Auscultation of the chest reveals inspiratory fine crepitations—described as “Velcro-like”. Chest auscultation is a fundamental investigation when suspecting IPF, as these changes are likely to be heard before changes become apparent on a plain chest radiograph. The presence of additional auscultatory findings, such as inspiratory squawks, should prompt evaluation for alternative diagnoses such as hypersensitivity pneumonitis [56].
High-resolution computed tomography (HRCT) is the key diagnostic test to reach the diagnosis of IPF. However, prior to any test a thorough clinical history is essential to (a) risk categorise a patient of being likely to have IPF (including a detailed occupational and family history), (b) determine exposures that might be causal for alternative causes of fILD, and (c) to exclude an underlying systemic disease which can cause similar CT changes (CTD features).
A focused history of autoimmune (e.g., CTD) features is required. Symptoms include Raynaud’s phenomenon, small joint arthropathy, sicca syndrome, skin changes, gastro-oesophageal reflux, and muscle pain/weakness. A history of exposure to bird feathers and mould or damp should also be established. It is common practice for CTD serology to be measured, including rheumatoid factor (RF), anti-nuclear antigen (ANA), extractable nuclear antigen (ENA), and myositis antibodies [57].
Should neither patient history nor serology be suggestive of an aetiological association with fILD, then IPF is likely to be the diagnosis if there are findings of subpleural, basal predominant, definite UIP pattern on HRCT [57]. A definite UIP will show features of honeycomb cystic change, in addition to traction bronchi- and bronchiolectasis. If there is an absence of honeycombing but the remaining features are found, the pattern will be designated a probable UIP. If the patient otherwise fits the expected profile (e.g., a male in his 7th decade), then this may be sufficient to reach the diagnosis (which requires multidisciplinary team (MDT) consensus).
In the situation where there is clinical uncertainty, then a tissue diagnosis may be necessary [58]. This can be via the use of Video Assisted Thoracoscopic Surgery (VATS), although the use of transbronchial cryobiopsy is an alternative option with a high diagnostic yield [59]. The histopathological findings of heterogenous dense interstitial fibrosis, fibroblastic foci, and honeycomb cysts are central to UIP. The absence of other features (granuloma and giant cells seen in hypersensitivity pneumonitis, and lymphoid aggregates and germinal centres seen in CTD-ILD) will support the diagnosis of IPF [60].
There is no role for routine bronchoscopy and bronchoalveolar lavage (BAL) in the diagnosis of IPF. However, BAL is appropriate when there are features that make an alternative diagnosis more likely, such as extensive ground glass opacification on CT.
Finally, pulmonary function tests (PFTs) help support the diagnosis and are critical for monitoring purposes. A restrictive defect is often found and supported by the presence of an FVC less than 80% of predicted for their height and age. The gas transfer factor for carbon monoxide (TLCO) is also likely to be low, reflective of the parenchymal disease impairing gas diffusion across the alveolar membrane.
No serological markers are currently clinically utilised. Krebs von den Lungen-6 (KL-6), the matrix metalloproteinases, and other markers of epithelial cell injury have been investigated but are currently only used in the research setting, although they may have a role for risk stratification and early diagnosis in future. KL-6, when elevated at the time of diagnosis or when rising during serial measurement, has been associated with accelerated lung function decline and poorer outcomes [61]. KL-6, therefore, has potential as a prognostic marker. MMP-1 and MMP-7 may be elevated in the serum of patients with IPF and may help distinguish IPF from other fILD, thus aiding diagnosis [62]. Baseline MMP-7 is associated both with progressive disease and overall mortality and therefore may have utility as a prognostic biomarker [63]. Surfactant proteins A and D may distinguish early disease from that of more advanced fibrosis [64] and therefore may be useful in guiding early intervention. However, all of these biomarkers are yet to be the subject of large-scale studies and their clinical utility remains to be determined.
Only two agents, pirfenidone and nintedanib, are currently licensed for treatment of fibrotic lung disease; however, a third drug nerandomilast, may be approved in the near future, having successfully met the primary endpoint in a very recent phase 3 trial (see “Future Directions” below). Pirfenidone may be initiated in a patient with IPF when the FVC is between 50% and 80% of predicted. This was previously true for nintedanib; however, NICE now approve this agent with no upper FVC limit [65].
Nintedanib is also licensed in the setting of non-IPF progressive fILD, whereas pirfenidone is not [66].
Pirfenidone’s mechanism of action is not clearly defined; however, several
anti-fibrotic properties have been demonstrated in vitro and in
vivo, including attenuation of TGF
Nintedanib is an inhibitor of multiple tyrosine kinases, including platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), and fibroblast growth factor receptor (FGFR) and is also licensed for use in lung cancer. In the phase 3 Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis (INPULSIS) trials, the rate of decline in FVC was halved in patients receiving nintedanib compared with placebo [71]. Diarrhoea is common, affecting almost two-thirds of patients. This can be managed with common anti-diarrhoeal agents such as loperamide; however, in persistent cases, a dose reduction may be necessary. As nintedanib acts via VEGFR, careful consideration should be given to patients with bleeding disorders. Although fewer patients stop nintedanib in the first three months, long-term adverse effects are common with approximately 58% of patients remaining on therapy at two years [72].
Both treatments are classed as antifibrotic therapies and not immunosuppressants. Treatment interruption is not necessary in the setting of infection, although in severe cases (sepsis/septic shock), this may be considered. Temporary suspension may be considered prior to surgery and during the treatment of pneumothorax, given the inhibitory effects of the agents on wound healing [73], which is particularly relevant with nintedanib therapy, given its action on vascular pathways. Pirfenidone may have a prophylactic role in preventing post-operative acute exacerbations following lung cancer resection in IPF patients [74].
For the treatment of ILD associated with CTD, there is good evidence of the use of the immunosuppressants, including cyclophosphamide and mycophenolate mofetil [75]. Similarly, steroids and immunosuppression are commonly prescribed for fILD, such as fHP, despite a limited evidence base [3, 76]. However, immunosuppression, including the use of corticosteroids and azathioprine, is contraindicated in the long-term management of IPF following the Prednisolone, Azathioprine, and N-acetylcysteine for Pulmonary Fibrosis (PANTHER-IPF) trial, which demonstrated increased mortality with immunomodulation [77]. Thus, it is fundamentally important to get an accurate diagnosis of IPF. However, high dose steroids may be considered in acute exacerbations of IPF on a case-by-case basis (see below).
Treatment of pulmonary hypertension (PH) is a particular challenge in IPF and there is conflicting evidence on the use of specific pulmonary vasodilators. Two RCTs—ARTERMIS-IPF and Riociguat for Idiopathic Interstitial Pneumonia-Associated Pulmonary Hypertension (RISE-IIP)—assessed the efficacy of ambrisentan and riociguat, respectively, in patients with IPF; however, both trials were terminated early due to increased risk of disease progression and adverse events [78, 79]. Conversely, treatment of severe forms of PH in IPF with sildenafil may help symptoms without worsening outcomes [80]. More recently, the prostacyclin analogue treprostinil has been shown to improve symptoms of PH but also displays potential antifibrotic properties (see Future Directions below). The management of PH in IPF must therefore be individualised and it must be borne in mind that certain specific treatments are contraindicated.
Symptomatic management in patients with IPF also requires careful consideration. Pulmonary rehabilitation should be considered for dyspnoea relief [81]. Oral modified-release morphine sulphate has recently shown benefit in cough management [82]. For patients with symptomatic Gastro Oesophageal Reflux Disease (GORD) proton pump inhibitors can be prescribed, although there is some evidence supporting a causal role for GORD in IPF [27], there is insufficient evidence to support routine use of proton pump inhibitors (PPIs) (or other antacid approaches) as disease modifying treatments. Ambulatory or long-term oxygen therapy may be appropriate in patients with exertional desaturation or type 1 respiratory failure, respectively.
Comprehensive health promotion should be extended to all patients with IPF. This includes critical measures such as smoking cessation support for current smokers and guidance on avoiding second-hand smoke for non-smokers. Additionally, participation in national vaccination programs against respiratory viruses should be strongly encouraged, particularly annual vaccinations against influenza and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), as infections with these viruses can have severe or even fatal consequences for IPF patients [83].
Lung transplant represents the only available treatment with proven survival benefit in IPF, and indeed, patients with fILD currently represent the largest proportion of patients on the transplant waiting list [84]. Although lung transplant is not a “cure”, it can significantly improve symptoms and quality of life. Median survival post-transplant is 6.7 years [85]. Allograft dysfunction and complications of immunosuppression, including a heightened risk of malignancy, account for these survival estimates [86]. The greatest risk of mortality is in the first year following transplant. Contraindications to transplant are listed in Table 2. An age of 65 is generally considered a cut-off for transplant in the UK; however, increasingly physiological age is factored more so than chronological age [87]. Transplant centres across the country are highly receptive to discussion and advocate for early referrals.
| Contraindications to lung transplant | |
| Absolute | Relative |
| Active malignancy | Age |
| Severe organ dysfunction (renal dysfunction with eGFR |
Organ dysfunction (ejection fraction |
| Recent MI or CVA | Gastro-oesophageal reflux disease |
| Sepsis or active acute infection | Osteoporosis |
| Substance dependence (including nicotine and alcohol) | Poor performance status |
| Poor adherence to medical therapies | Extremes of weight (BMI |
This list is not exhaustive. Relative contraindications should be considered on an individual basis and a patient’s condition optimised where possible. BMI, body mass index; CVA, cerebrovascular accident; eGFR, estimated glomerular filtration rate; MI, myocardial infarction.
An acute exacerbation of IPF (AE-IPF), defined as a worsening of respiratory symptoms with the presence of new bilateral ground glass or consolidation on CT not caused by cardiac failure, is associated with high mortality [88]. In-hospital mortality may be as high as 50%. After exclusion of other reversible causes, such as pulmonary embolism and pneumothorax, patients should be screened for a treatable infective cause, including respiratory viral pathogens by PCR and sputum microscopy, cultures, and sensitivity testing. Procalcitonin may guide the need for antibiotic treatment. Bronchoalveolar lavage is usually not necessary or feasible. Atypical pathogens should be considered, and testing may include a serum beta-D-glucan to evaluate for Pneumocystis jirovecii as well as cytomegalovirus (CMV) serology (albeit less likely causes of respiratory deterioration given IPF patients are seldom immunosuppressed).
Where appropriate, such patients should be discussed with the intensive care team and their transplant team without delay if they have already been assessed. Oxygenation (including the use of high-flow nasal oxygen, continuous positive airway pressure (CPAP), and non invasive ventilation (NIV)) should be prioritised, even if for symptomatic relief. Use of NIV may offer some benefit, with one small study showing that the use of NIV may avoid the need for endotracheal intubation which otherwise has unambiguously poor outcomes [89, 90], RCT data are, however, lacking.
The administration of high dose pulse intravenous steroids (e.g., methylprednisolone) may be considered as a “rescue therapy” and remains a weak recommendation from the American Thoracic Society (ATS) guidelines with a conflicting evidence base. Evidence is largely from observational studies with one cohort study demonstrating a survival benefit of high dose methylprednisolone followed by oral prednisolone if no poor prognostic factors (including the use of positive pressure ventilation) were present [91]. However, a U.S. retrospective analysis found no improved survival benefit when corticosteroids were administered for AE-IPF [92] which is supported by a recent meta-analysis, which found no survival benefit in AE-IPF but supports a potential survival benefit when high dose corticosteroids are administered in non-IPF fILD [93]. There is good evidence against escalating immunosuppression beyond steroid therapy with an RCT demonstrating increased mortality when cyclophosphamide is administered alongside steroid therapy [94].
Steroid therapy should therefore be used on a case-by-case basis, due to the limited evidence base. However, given the huge risk of deterioration that patients with AE-IPF face and the potential inflammatory contribution to pathogenesis, steroids are frequently utilised.
Early involvement of palliative care teams is often appropriate.
Since the licensing of pirfenidone and nintedanib in 2014, no new therapies have been licensed for use in IPF to date. This disappointing milestone is hopefully about to change, as the phosphodiesterase 4B (PDE4B) inhibitor nerandomilast has recently completed phase 3 trials and met its primary endpoint of change in FVC at 52 weeks, according to the company press release [95]. Full results of the trial are expected in 2025 and the company are proceeding with licensing approvals.
Inhaled treprostinil has been shown to improve symptoms and is well tolerated, specifically in patients with PH ILD [96]. Interestingly patients also demonstrated stability in FVC (which was monitored for safety purposes) suggesting this vascular targeted treatment may indeed have antifibrotic properties. The TETON trial using inhaled treprostinil in IPF subjects with change in FVC as the primary endpoint is fully recruited and data are expected in 2025 [97]. Disappointingly, a phase 2b/3 runthrough trial assessing efficacy of the novel dual integrin inhibitor bexotegrast has recently been paused following an interim data review [98]. Similar discouraging results have been observed with an lysophosphatidic acid receptor 1 (LPAR1) antagonist fipaxalparant, although a similar compound admilparant has shown more promise and is recruiting for a phase 3 trial [99]. Other trials in progress are shown in Table 3.
| Compound | Mechanism | Trial details with clinicaltrials.gov reference |
| Admilparant | LPAR1 antagonist | Phase 3; NCT06003426 |
| Buloxibutid | Angiotensin II type receptor agonist | Phase 3; NCT06588686 |
| Garadacimab | Monoclonal antibody targeting FXIIa | Phase 2a; NCT05130970 |
| Zelasudil | Rho kinase II inhibitor | Phase 2; NCT05570058 |
| Taladegib | Hedgehog pathway inhibitor | Phase 2; NCT06422884 |
| Leramistat | NADH dehydrogenase inhibitor | Phase 2; NCT05951296 |
FXIIa, activated factor XII; LPAR1, lysophosphatidic acid receptor 1; NADH, nicotinamide adenine dinucleotide hydride.
Several challenges stand in the way of recruiting for IPF clinical trials including patient age and the short life expectancy and the relatively small pool of patients who may meet likely strict inclusion criteria. In addition, the pharmaceutical development process is often prohibitively protracted. Strategies to overcome these barriers by embedding clinical trials into routine clinical care and the use of adaptive platform trials present a novel method of expanding therapies in IPF. The multinational “Randomised, Embedded, Multifactorial Adaptive Platform Trial for Interstitial Lung Disease (REMAP-ILD)” effort represents a drive to develop novel and repurposed approaches for patients suffering from IPF [100].
IPF remains a challenging condition to manage. Our understanding of the disease process, risk factors and triggers is growing and therapeutic progress is being made. However, the rising incidence and generally poor outcomes remain a cause for considerable concern. An individualised approach to all such patients is vital and an awareness of a patient’s advanced care plan and transplant status will help the clinician in formulating a management plan. With novel treatments proving efficacious and a plethora of clinical trials underway, there does remain much promise that outcomes will improve for these patients and that a cure for fibrosis is on the horizon.
All the data of this study are included in this article.
Both JM and GJ contributed to the concept and design of the manuscript. JM drafted the manuscript, made subsequent revisions, and made both original figures. GJ made substantial revisions and provided senior oversight in its production. Both JM and GJ have approved the final version of the manuscript for publication and are accountable for all aspects of the work.
Not applicable.
Not applicable.
James May receives funding from the Medical Research Council (MRC) via a Clinical Research Training Fellowship (MR/X001814/1). Gisli Jenkins is funded by a National Institute for Health and Care Research (NIHR) Research Professorship (RP-2017-08-ST2-014) and receives funding from the MRC supporting related work from grants MR/V00235X/1, MR/W014491/1, and MR/W031469/1.
James May declares no conflict of interest. Gisli Jenkins reports honoraria from Boehringer Ingelheim, Chiesi, Roche, PaZentMPower, AstraZeneca, GSK; and consulting fees from AbbVie, AdALta, Apollo TherapeuZcs, Brainomix, Bristol Myers Squibb, Chiesi, Cohbar, GlaxoSmithKline Pliant, RedX. Figs. 1,2 were created using BioRender.com. The authors have no financial or personal relationship with BioRender.com, and the use of this tool does not imply any endorsement.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.