
The function of the fibroblast growth factor receptor 3 (FGFR3) gene in bone development was first demonstrated by Shiang et al. [1], who reported an FGFR3 mutation in patients with achondroplasia. The field of bone biology was significantly advanced by that discovery. Other reports followed, and a series of FGFR3-related skeletal dysplasias such as thanatophoric dysplasia (TD) and hypochondroplasia were associated with mutations in the FGFR3 gene. TD is the most common form of neonatal lethal skeletal dysplasia with an estimated frequency of 1: 20,000 to 1: 12,000 in prenatal cases [2]. TD is a sporadic, autosomal-dominant skeletal dysplasia that is classified as TD type I (TD1) and TD type II (TD2), based on the clinical features. Recently, different FGFR3 mutations were linked to either subtype. TD1 is caused by missense or stop codon mutations in the FGFR3 gene, whereas TD2 is only known to be caused by the FGFR3 p. Lys650Glu missense mutation [3]. Pathogenic allelic variants in FGFR3 have been associated with many different phenotypes. A correlation between genotype and phenotype has not been apparent due to the limited data regarding FGFR3-related skeletal dysplasia. In the study, the authors investigated the FGFR3-mutation genotype in eight fetuses with TD1, which were diagnosed by prenatal ultrasound evaluation and postnatal radiographic findings. The aim of this study was to provide additional data for genetic analysis of TD1 with FGFR3 mutations.
The Prenatal Diagnosis Center of Xiamen Maternal and Child Health Care Hospital is a regional tertiary referral center for expectant mothers whose fetuses have suspected anomalies and/or genetic syndromes. The center provides prenatal services for a considerable percentage of suspected anomalous pregnancies in the southwest area of Fujian Province in mainland China. In China, pregnancy can be terminated in any trimester if the fetus has severe malformations. Informed consent was obtained from parents of the fetuses. The study was approved by the Ethics Committee of Xiamen Maternal and Child Health Care Hospital.
Eight cases received of TD1 diagnosed by prenatal ultrasound evaluation and postnatal radiographic findings between January 2015 and June 2017. These cases of severe, fetal shortlimb dwarfism detected by antenatal ultrasonography were referred to this center. The women involved were in non-consanguineous marriages and had a normal course of pregnancy; their family histories were devoid of other reports of skeletal malformations. Because the prenatal ultrasonography findings of these cases were predicted to have potential lethal outcomes, the families decided to terminate the pregnancies. Before each pregnancy was terminated, cordocentesis was performed for fetal karyotyping and molecular analysis. Postmortem radiographic examination and gross autopsy were performed in each instance for definitive diagnosis.
In this study, prenatal ultrasound examination of the eight fetuses revealed marked shortening, bowed limbs, a narrow thorax, a protuberant abdomen, and polyhydramnios, so TD1 was suspected. Postmortem radiological features showed very short, curved long bones, short ribs without fractures, severe platyspondyly, decreased interpediculate distances in the region of the lumber spine, and without a clover-leaf skull. TD1 was confirmed based on these findings [4].
Genomic DNA was extracted using DNA blood kit, according to the manufacturer’s standard procedure. The genomic DNA was fragmented using an LE220 focused ultrasonicator to generate a paired-end library (200-250 bp). Using a 2100 bioanalyzer and a real-time PCR System, the magnitude of enrichment was estimated. After quality control, the captured library was sequenced using an analyzer for 90 cycles per read to generate paired-end reads. Image analysis, error estimation, and base calling were performed using Illumina Pipeline software (version 1.3.4). After bioinformatics processing, the sequences were aligned to human genome reference sequence hg19, using the Burrows Wheeler Aligner Multi-Vision software package. SOAPsnp and SAMtools were used to detect singlenucleotide variants and insertions and deletions (indels). All single-nucleotide variants and indels were filtered and searched against multiple databases, including NCBI dbSNP, HapMap, the 1000 Human Genomes Project dataset, and the database of 100 Chinese healthy adults [5]. All mutations and potential pathogenic variants were validated by conventional Sanger sequencing.
Typical ultrasound findings, such as marked shortening, bowed limbs, a narrow thorax, and a protuberant abdomen were found in all cases, and polyhydramnios were found in two cases. Sonographic measurements demonstrated a fetal femur and/or humerus length that was > 4 standard deviations below the mean femoral length (FL) and/or humeral length (HL) for the gestational age, as predicted by Hadlock et al. [6]. The results of the ultrasound measurements for each of the eight cases are shown in Table 1.
Differential radiological features such as very short, curved long bones, short ribs without fracture, severe platyspondyly, and normal skull without a clover-leaf shape were observed in all cases, which is consistent with the diagnosis of TD1. The radiographic pictures of the eight cases are presented in Figure 1.
| Case | Maternal age (y) | Gestational age (weeks) | Ultrasound findings | Radiographic diagnosis | Molecular analysis results |
|---|---|---|---|---|---|
| 1 | 28 | 26 + 2 | BPD: 7.2 cm, FL: 1.9 cm, HL: 1.7 cm | TD1, Figure 1(a) | c.1118A>G, p.Tyr373Cys |
| 2 | 27 | 26 | BPD: 7.8 cm, FL: 2.3 cm, HL: 2.1 cm | TD1, Figure 1(b) | c.742C>T, p.Arg248Cys |
| 3 | 25 | 23 + 1 | BPD: 5.7 cm, FL: 1.8 cm, HL: 1.8 cm | TD1, Figure 1(c) | c.742C>T, p.Arg248Cys |
| 4 | 34 | 25 + 3 | BPD, 7.6 cm: FL, 1.9 cm: HL, 2.0 cm | TD1, Figure 1(d) | c.742C>T, p.Arg248Cys |
| 5 | 26 | 24 + 2 | BPD: 6.0 cm, FL: 1.9 cm, HL: 2.0 cm | TD1, Figure 1(e) | c.2419T>G, p.Ter807Gly |
| 6 | 38 | 25 + 4 | BPD: 6.8 cm, FL: 2.0 cm, HL: 2.0 cm | TD1, Figure 1(f) | c.2421A>C, p.Ter807Cys |
| 7 | 33 | 24 | BPD: 5.9 cm, FL: 1.7 cm, HL: 1.7 cm | TD1, Figure 1(g) | c.742C>T, p.Arg248Cys |
| 8 | 34 | 25 + 2 | BPD: 6.4 cm, FL: 1.9 cm, HL: 2.0 cm | TD1, Figure 1(h) | c.1043C>G, p.Ser348Cys |
BPD: biparietal diameter; FL: femoral length; HL: humeral length.
Distinct heterozygous mutations in the FGFR3 gene were identified in the eight fetuses by FGFR3 gene sequencing. Among the FGFR3 mutations identified, six cases had missense mutations corresponding to the extracellular domain of the encoded FGFR3 protein, and two cases had stop codon mutations. Of the two cases with stop codon mutations, one case had a heterozygous c.2419T>G (p.Ter807Gly) mutation, and the other case had a heterozygous c.2421A>C (p.Ter807Cys) mutation. Among the six missense mutations identified in the FGFR3 gene, four cases had a heterozygous c.742>T (p.Arg248Cys) mutation, one case had a heterozygous c.1118A>G (p.Tyr373Cys) mutation, and one case had a heterozygous c.1043C>G (p.Ser348Cys) mutation, which has been reported previously in achondroplasia and hypochondroplasia with acanthosis nigricans [7, 8]. This is the first report of a missense FGFR3 p.Ser348Cys mutation identified in a fetus with TD1. The molecular-analysis results for the FGFR3 gene in the eight cases are shown in Table 1. The fetal karyotypes of the eight fetuses were normal.
Gross autopsies revealed that the eight fetuses had macrocephaly, a depressed nasal bridge, micromelia, a small bell-shaped thorax, and a protruding abdomen. Extra malformations were found in three cases, among which cerebral dysgenesis and ureterostenosis were found in case 5, hydronephrosis was found in case 6, and the absence of the right testis was found in case 8.
TD is characterized by marked shortening of the limbs, a narrow thorax, macrocephaly with frontal bossing, and a relatively normal trunk length [9]. Affected individuals usually die soon after birth. Because of its lethality, prenatal diagnosis of TD is performed by ultrasonography in the second trimester. Sonographic features associated with lethality include marked shortening, bowed limbs, narrow thorax, a protuberant abdomen, and polyhydramnios [10]. Additional malformations such as central nervous system, renal, and cardiovascular anomalies may also be involved and can be detected by ultrasound and autopsy. However, the lethality of skeletal dysplasia can be recognized during the antenatal period with confidence, the diagnosis of a special skeletal dysplasia is challenging and often made postmortem with the assistance of a complete skeletal survey. The diagnostic accuracy can be improved to almost 80% by analyzing fetuses for known radiographic characteristics in individual with skeletal dysplasia [11]. Molecular techniques have gradually become the only available methods to confirm the diagnosis of skeletal dysplasia.
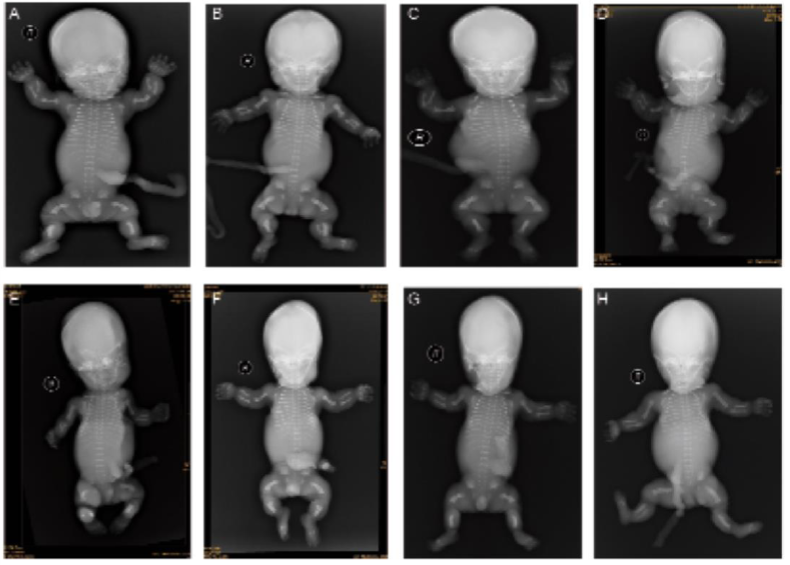 Figure 1.
Figure 1.— Differential radiological features such as very short, curved long bones, short ribs without fracture, severe platyspondyly, and a normal skull without a clover-leaf shape are present in all cases, which is consistent with the diagnosis of TD1.
TD has been subdivided into two categories based on the presence or absence of a clover-leaf skull and the presence of curved, rather than straight femurs [12]. TD1 is characterized by curved femurs (telephone receiver appearance), usually without a clover-leaf skull. In contrast, patients with TD2 exhibit relatively straight femurs associated with a clover-leaf skull. In 1988, Spranger classified hypochondroplasia (HCH), achondroplasia (ACH), and thanatophoric dysplasias (TD1 and TD2) as belonging to the same family of dysplasias [13], because these dysplasias (which may result from the same genetic pathology) display common phenotypic characteristics with different grades of severity. In 1994, Spranger’s clinical observations were subsequently confirmed by the discovery that functionally activating mutations in FGFR3 account for the common genetic form of dwarfism in human achondroplasia. To date, many findings have shown that ACH, HCH, TD1, and TD2 are caused by several different mutations in the FGFR3 gene [14].
The FGFR3 gene is a transmembrane tyrosine kinase receptor that binds fibroblast growth factors. The FGFR3 gene, isolated in 1991, is located on chromosome 4 (4p16.3) [15] and is comprised of 19 exons and 18 introns [16]. The FGFR3 protein comprises three main parts: an extracellular region including three immunoglobulin (Ig) domains, a transmembrane segment, and an intracellular region comprising two tyrosine kinase (TK) domains [17]. Mutations in FGFR3 causing skeletal dysplasias are all inherited in an autosomal dominant pattern, but frequently occur de novo on the paternal allele. Experimental studies often demonstrate that mutations activate FGFR3 in a constitutive, ligand-independent fashion and that the degree of activation correlates directly with the severity of the associated skeletal phenotype [18]. TD1 is likely to manifest clinically from a genetically homogeneous mutation at the FGFR3 locus. Several amino acid substitutions in the extracellular linker region between the immunoglobulin-like domains in the FGFR3 protein have been found in TD1, including p.Arg248Cys and p.Tyr373Cys [19-21]. In addition, several stop codon mutations have also been described [19-21]. Only the p.Lys650Glu mutation detected in the tyrosine kinase II (TDII) domain of FGFR3 has been found in TD2 [12]. ACH largel accounted for a single p.Gly380Arg mutation corresponding to an amino acid in the transmembrane domain of FGFR3 [22]. Mutations causing HCH are more widespread in FGFR3 with a hotspot in the tyrosine kinase domain at codon 540 in exon 13 [23].
In this study, the c.742>T (p.Arg248Cys) mutation in the FGFR3 gene showed the highest frequency, accounting for 50% of all TD1 cases. Another missense mutation (c.1118A>G; p.Tyr373Cys) was identified in one case, and the other 25% of cases were accounted for by stop codon mutations and a rare FGFR3 mutation at position 1043 (p.Ser348Cys), found in one case. Ser348 resides in the third immunoglobulin-like domain of FGFR3 gene near other mutations associated with TD and ACH. Ser 348 in FGFR3 is highly conserved in humans. Using analysis tools to predict the functional effects of amino acid substitutions, p.Ser348Cys was determined to be probably damaging with PolyPhen2 [24] score of 0.981 and was predicted to be disease causing with Mutation Taster [25] score of 112. The p.Ser348Cys mutation in FGFR3 has been reported to cause achondroplasia and hypochondroplasia with acanthosis nigricans [7, 8]. The present results revealed the first TD1 case that was diagnosed by prenatal ultrasound evaluation and postnatal radiographic findings, with a heterozygous c.1043C>G (p.Ser348Cys) mutation in the FGFR3 gene. The present authors conclude that this mutation was likely responsible for the clinical and radiological features of FGFR3-related skeletal dysplasias. The findings of this study add to the extensive variability in clinical disease presentation and reveal molecular defects in patients with FGFR3 mutations.
The present data confirm the existence of hotspot mutations in TD1 and suggest that a considerable overlap may exist in the genotypes and phenotypes in ACH, HCH, TD1, and TD2. Mutations in the FGFR3 gene show wide clinical variability in individuals with FGFR3-related skeletal dysplasias. As far as genotype-phenotype correlations are concerned, no substantial X-ray differences among TD1 patients were noted, regardless of the location of the mutation in the FGFR3 gene [21]. Advancements in molecular techniques have resulted in substantial progress in identifying molecular defects responsible for FGFR3-related skeletal dysplasias. Genotype-phenotype correlations are becoming increasing pertinent with the continual increase in the number of mutations identified in FGFR3-related skeletal dysplasias. The increasing accumulation of associations between mutations and clinical data is an important component of genetic counseling and sheds insight into etiological factors of FGFR3-related skeletal dysplasias for evaluating and refining current models relating genotype to phenotype.
This study was supported by the Science and Technology Committee of Fujian Province [grant number 2015-CXB-44].