
Objective: In this article we present a case of fetal nemaline myopathy (NM) diagnosed by whole-exome sequencing (WES) and confirmed by fetal muscular pathology, and we review the clinical, pathological, and genetic characteristics of congenital NM. Method: A pregnant woman with recurrent fetal hydrops and polyhydramnios was recommended to undergo WES before termination of pregnancy in order to find the etiology of these issues. After delivery, we obtained umbilical-cord blood and parental peripheral blood samples for WES. Fetal muscle was subjected to modified Gomori technique (MGT) and hematoxylin phosphotungstate (PTAH) staining for light microscope detection. Results: WES revealed two compound heterozygous mutations to fetal Kelch-like 40 (KLHL40), and pedigree-based Sanger sequencing showed that c.602G > A (p.Trp201*) was inherited from the mother and c.1516A > C (p.Thr506Pro) from the father. MGT and PTAH staining highlighted numerous nemaline rods under the light microscope. Conclusion: Fetal NM is a lethal genetic muscle disorder that is one etiology of fetal akinesia deformation sequence (FADS). Pathological and genetic testing are the current diagnostic methods for NM, but WES is a promising method for prenatal diagnosis of this disorder.
Nemaline myopathy (NM) includes a clinically and genetically heterogeneous group of congenital muscle disorders, with age of onset varying from newborn to elderly [1]. At the most severe end of the NM clinical spectrum are genetically unresolved cases of autosomal-recessive fetal akinesia deformation sequence (FADS) [2], a term covering a broad spectrum of disorders whose unifying feature is a reduction in or lack of fetal movement [3]. Associated abnormalities can include arthrogryposis, pterygia, subcutaneous edema, fetal hydrops, lung hypoplasia, rocker-bottom feet, craniofacial anomalies (particularly cleft palate and retromicrognathia), intrauterine-growth retardation, and poor muscle bulk [4]. Fetal NM, the most serious form of NM that leads to FADS, is typically lethal in the fetal or neonatal period. Even with a multidisciplinary approach involving collaborations among specialists in prenatal ultrasonographic (US) imaging, clinical genetics, and fetal pathology, a definitive diagnosis can be made in no more than half of cases [5]. Herein, we report a case of intrauterine-onset fetal NM diagnosed via WES and confirmed by fetal muscular pathology. We reviewed the clinical, pathological, and genetic characteristics of NM along with the case report to improve our understanding of the disorder, as well as to provide more-cogent empirical evidence that might direct targeted genetic counseling and prenatal diagnosis of this disorder in the future.
A 31-year-old woman, G3P1, in a non-consanguineous marriage exhibited normal physical and mental development. Both she and her husband denied any family history of hereditary disease. In 2010, she delivered a full-term, live, healthy male infant. During her second pregnancy in 2016, she underwent induced labor at 34 weeks of gestation due to fetal death with hydropic appearance and polyhydramnios; she also developed postpartum hemorrhaging with possible amniotic-fluid embolism but recovered after resuscitations. The current pregnancy was her third. Although US screening at 23 weeks did not show any fetal malformations, the mother had not felt any fetal movements during this pregnancy, similar to her second pregnancy. At 32+4 weeks, US showed abnormal fetal left-finger posture, thickened subcutaneous soft tissue in the thoracic and abdominal areas, and borderline polyhydramnios. The woman was transferred to our hospital at 33+5 weeks. Prenatal US showed a single live intrauterine fetus, with thickened whole-body skin (scalp thickness, 0.7 cm; trunk skin thickness, 0.6 cm), fixed extremities, overlapping fingers, and polyhydramnios that presented with the same abnormal US findings as her second pregnancy. Because the features detected by antenatal US were focused on neonatal or infantile lethality, the family decided to terminate the pregnancy. After a multidisciplinary consultation with prenatal specialists, whole-exome sequencing (WES) was recommended in order to determine etiology before termination. Because of abdominal bloating and difficulty lying down due to polyhydramnios, the patient underwent amniocentesis that drained approximately 3,000 mL amniotic fluid. She received an intravenous infusion of 0.5% oxytocin to induce labor on the day after amniocentesis; labor progressed uneventfully. After the cervix was almost fully dilated, an incision was made high in the amniotic membrane, and about 2,000 mL amniotic fluid was released. A live baby girl was delivered with a birth weight of 2,145 g, Apgar scores of 4 → 2 → 1 points, and post-delivery amniotic-fluid volume of 3,000 mL. Physical examination showed a neonate with generalized skin edema, laryngeal edema, and fixed and rigid extremity posture with no muscle tone. The patient chose to discontinue resuscitation of the infant after discussion with her family. We obtained umbilical-cord and parental peripheral blood samples (5 mL each) and sent them for WES, and the newborn was sent for postmortem pathological examination.
Whole-exome sequencing. We extracted genomic deoxyribonucleic acid (DNA) from the blood samples using a MagPure Buffy Coat DNA Midi KF Kit. A single individual DNA library was built after fragmentation, ligation-mediated polymerase chain reaction (LM-PCR), and purification. The library was enriched for 16-24 h at 47 ℃ by array hybridization (Roche NimbleGen, Madison, WI, US), followed by elution and post-capture amplification. The products were then analyzed with Agilent 2100 Bioanalyzer and BMG to estimate the magnitude of enrichment. The qualified products were pooled and quantified according different library quantities, then the singlestrand of library products were prepared for circularization and made DNB, finally, sequenced with PE100 + 100 on MGISEQ-2000. After sequencing was completed, we obtained the original sequence data and aligned them using HG19/HG20 using Burrows-Wheeler Aligner (BWA) software (https://github.com/lh3/bwa), simultaneously evaluating the sequence capture effects. We assessed single-nucleotide variants (SNVs) and insertion and deletion (indel) using Genome Analysis Toolkit (GATK) software [6] in order to generate targeted polymorphic bases. These results were compared against the dbSNP database (National Institutes of Health [NIH], Bethesda, MD, US), the International HapMap Project (HapMap) database (National Center for Biotechnology Information [NCBI], Bethesda, MD, US), the 1000 Genomes Project dataset (http://www.internationalgenome.org/) and a database of 100 healthy Chinese adults to annotate and screen for suspicious mutations. All mutations and potential pathogenic variants were validated using conventional Sanger sequencing methods. To predict the effect of missense variants, we used the dbNSFP database (http://sites.google.com/site/jpopgen/dbNSFP), which contains several well-established software programs for in silico prediction (Scale-Invariant Feature Transform [SIFT], Polyphen2, Likelihood Ratio Test [LRT], MutationTaster and PhyloP). The Human Gene Mutation Database (HGMD) was used to screen mutations reported in published studies. We compared our results against the standard sequence of Kelch repeat and Bos taurus (Bovine) domain containing protein 5 (KBTBD5; NM-152393.2) to verify the results of gene chip capture and high-throughput sequencing (HTS).
Muscle biopsy examination. After we stained the fetal abdominal-muscle tissue using the modified Gomori technique (MGT) or hematoxylin phosphotungstate (PTAH), we examined the derived paraffin sections under a light microscope.
For MGT, we rinsed dewaxed paraffin sections in water. After oxidization with potassium permanganate for 5 min and bleaching with 2% oxalic acid, the sections were immersed in Gomori solution for 20-40 min, rinsed under running water, dehydrated, cleared, and mounted.
For PTAH (the natural oxidation method), we rinsed dewaxed paraffin sections in water, immersed them in freshly prepared PTAH oxidant for 5 min, and washed and bleached them with oxalic acid. The sections were soaked in Mallory PTAH staining solution for 24-48 h, and then dehydrated, cleared, and sealed.
The patient reported no fetal movements during this pregnancy, which had also occurred in her second pregnancy. Prenatal US showed thickened fetal skin (Figure 1), fixed-extremity posture, overlapping fingers (Figure 2), and polyhydramnios at 33+5 weeks of gestation. Amniocentesis was performed 2 days before termination of pregnancy, at which time we drained approximately 3,000 mL amniotic fluid; the total amount of amniotic fluid was estimated to be about 5,000 mL during delivery. Examination of the fetus after delivery revealed generalized skin edema, laryngeal edema, fixed and rigid extremities, and no muscle tone.
 Figure 1.
Figure 1.— Ultrasonographic examination shows fetal scalp thickness reaching 0.7 cm and trunk skin thickness reaching 0.6 cm.
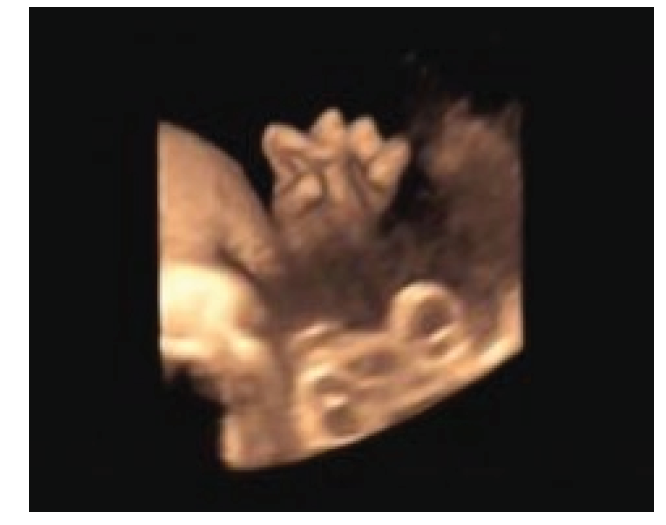 Figure 2.
Figure 2.— Overlapping fingers in the fetus under ultrasonographic examination.
MGT staining revealed a large number of bluish-purple rods in the subsarcolemma or sarcolemma of the muscle fibers (Figure 3). PTAH staining showed numerous blue rods (Figure 4).
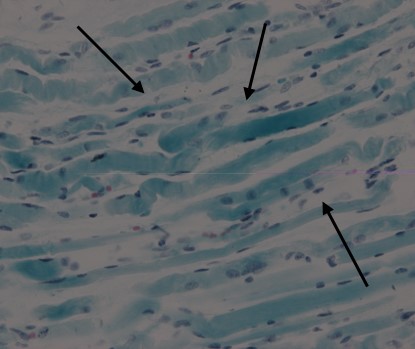 Figure 3.
Figure 3.— MGT staining shows a large number bluish-purple rods (400 ×) (arrows).
 Figure 4.
Figure 4.— Rods stained blue with PTAH staining (400 ×) (arrows).
WES of umbilical-cord blood revealed two complex heterozygous mutations to fetal Kelch-like 40 (KLHL40). Pedigree-based Sanger sequencing confirmed that the mutation c.602G > A (p.Trp201*) was inherited from the mother (Figure 5) and c.1516A > C (p.Thr506Pro) from the father (Figure 6).
 Figure 5.
Figure 5.— Sequencing map of exon 1 of the fetal and parental KLHL40 gene c.602G > A (p.Trp201*).
 Figure 6.
Figure 6.— Sequencing map of exon 4 of the fetal and parental KLHL40 gene c.1516A > C (p.Thr506Pro).
NM was first reported by Gonen et al. and Shy et al. in 1963 and was named for the large number of nemaline bodies or rods found in the muscle fibers of affected patients [7]. NM is a congenital myopathy that can present as an autosomal-dominant or -recessive disease or a sporadic disease, with an incidence of about 2 in 100,000 people [8]. In 1999, based on age at disease onset and severity of clinical manifestations, the European Neuromuscular Disease Center (ENMC) classified the disease into six subtypes: severe congenital NM, intermediate congenital NM, typical congenital NM, mild childhood or juvenile-onset NM, adult-onset NM, and other forms of NM [9]. At this writing, NM is a well-recognized myopathy involving multiple genetic defects and encompassing a broad clinical spectrum ranging from mild to severe muscle weakness [10]. At the most severe end of this spectrum are genetically unresolved cases of autosomal-recessive FADS [2], a term covering a broad spectrum of diseases characterized by diminished or lack of fetal movement in the uterus [11]. Other related prenatal features include fetal hydrops, arthrogryposis, pterygia, and polyhydramnios. Fetal akinesias are clinically and genetically heterogeneous, with causative mutations identified to date in a large number of genes encoding disparate parts of the motor system [12]. Fetal akinesias can be caused by defects at any point along the motor system pathway, including the central and peripheral nervous systems, neuromuscular junctions and muscle, as well as by restrictive dermopathy or external restriction of the fetus in utero [13]. Fetal NM, a lethal genetic muscle disorder, is one etiology of FADS.
The hallmark of all forms of NM, regardless of genetic defect, disease severity, or age of onset, is the presence of intrasarcoplasmic rod-like structures called nemaline bodies/rods, which are stained red by MGT or blue by PTAH [14]. Electron microscopy shows different lengths of rod-shaped bodies (with the same electron density as the z-lines of striated muscle) scattered among the myofibrils, with their long axes perpendicular to the z-line [1, 7]. Severely affected patients can have fatty infiltration and fibrous-tissue hyperplasia. In the past, diagnosis mainly relied on skeletal-muscle biopsy. However, prenatal diagnosis that relies on fetal muscle biopsy is not feasible, and with the development and application of molecular biotechnology, muscle biopsy is no longer the sole gold standard. The recent advance of WES can effectively provide a definitive molecular diagnosis. WES facilitates genetic diagnosis of fetal structural anomalies, which enables fetal prognosis and risk of recurrence to be more accurately predicted in future pregnancies [15]. An increasing number of NM cases have been reported based on diagnoses made via molecular-biology techniques.
NM is a heterogeneous group of genetic diseases. At this writing, mutations in more than 10 genes have been found to cause NM. Dominant mutations include those of actin alpha 1 (ACTA1), tropomyosins 3 and 2 (TPM3, TPM2) and KBTBD13; recessive mutations include those of nebulin (NEB), ACTA1, TPM3, TPM2, troponin T1 (TNNT1), cofilin 2 (CFL2), KLHL40, KLHL41, leiomodin-3 (LMOD3), myopalladin (MYPN) and myosin-XVIIIb (MYO18B) [16, 17]. NEB mutation is the most common cause of NM and is responsible for about 50% of the mutations identified, while ACTA1 is the second most common pathogenic gene in NM, accounting for about 15%-25% of mutations [16, 17]. Some studies have demonstrated that KLHL40 mutations cause a significant proportion of severe NM cases of FADS, and the disease shows worldwide prevalence [18, 19].
Ravenscroft et al. [19] studied a multinational cohort of 143 families affected by severe congenital NM who had not been genetically diagnosed. After performing WES of six families and targeted gene sequencing of additional families, they identified 19 mutations to KLHL40 in 28 apparently unrelated NM kindreds of various ethnicities. Accounting for up to 28% of the tested individuals in the Japanese cohort, KLHL40 mutations were found to be the most common cause of this severe form of NM. Clinical features of affected individuals were distinctive and included fetal akinesia or hypokinesia and contractures, fractures, respiratory failure, and swallowing difficulties at birth. Ravenscroft’s team also screened 129 probands with milder NM but found no KLHL40 mutations in this cohort, confirming that such mutations are most likely exclusive to cases of severe NM. Expression of KLHL40 in fetal and adult skeletal muscle indicates that KLHL40 plays a role in both myogenesis and mature muscle and probably explains why mutation to this gene is the most common cause of the severest form of NM associated with FADS [20].
In our study, the pregnant woman with recurrent fetal hydrops and polyhydramnios was recommended to undergo WES to clarify the etiology of fetal deformity. WES revealed two compound heterozygous mutations, c.602G > A (p.Trp201*) and c.1516A > C (p.Thr506Pro), to KLHL40; these have been reported previously as pathogenic-mutation sites. Based on the genetic information and clinical features of the case, we diagnosed fetal NM consistent with FADS. Fetal-muscle biopsy, the diagnostic gold standard, showed typical rods after MGT or PTAH staining, which confirmed the diagnosis of intrauterine-onset fetal NM. Recently, delineation of fetal-anomaly phenotypes has received increasing attention, including a discussion of whether to introduce WES in standard prenatal care, particularly for families with one or more fetuses presenting a distinctive anomaly pattern and/or phenotype recurrence with increased risk of lethal outcome. The present study demonstrated that WES has the potential to improve clinical management of pregnancies and better inform the reproductive decisions of affected families. Therefore, WES is a promising method for prenatal diagnosis of NM. With the rapid development of gene detection technology, genes related to hereditary neuromuscular diseases and new mutation sites of genes have been discovered. These new findings provide a reliable basis for genetic diagnosis and prenatal detection of hereditary neuromuscular diseases.
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Women and Children’s Hospital Affiliated to Xiamen University (approval number: KY-021).
This project was supported by The Science and Technology Project of Xiamen, Fujian, P.R. China (Grant No.: 3502Z20174018).
The authors declare no conflict of interest.