
Academic Editor: Giovanni Monni
Background: We aimed to analyze mutations of the pathogenic gene in
dyssegmental dysplasia Silverman-Handmaker (DDSH) type associated with the
Heparin sulfate proteoglycan 2 (HSPG2) gene.
Case: Prenatal testing for genetic mutations associated with
fetal DDSH were performed on a pregnant woman with previous history of carrying a
fetus with short limb malformation at the 17th week of gestation. DNA was
extracted from amniotic fluid and next-generation sequencing-based deep panel
sequencing was performed on the Illumina NextSeq platform to identify possible
causative mutations of DDSH. Results: Two novel heterozygous
mutations in HSPG2 gene, c.6001dupC (p. R2001pfs*19) and c.11207G
Fetal short limb deformity is one of the most common congenital birth defects. While its etiology is diversified, it is primarily related to exposure to drugs, radiation, decoration pollution, chemicals such as hair dyes, benzene, mercury, lead and other heavy metals during pregnancy. Several trials point to recessive genetic diseases of chromosome [1, 2]. Fetal short limb deformities include fatal and non-fatal cases [3]. Related Ultrasound screening is mainly carried out in the second and third trimesters of pregnancy. However, for non-fatal skeletal dysplasia with atypical ultrasonographic features, it is difficult to make a definite diagnosis and classification, resulting in a high probability of misdiagnosis and missed diagnosis. Such failure in prenatal screening can result in disability due to skeletal deformity after birth and even complications of the nervous system that threaten great mental and economic burden to family and society [4, 5, 6, 7]. Accordingly, timely and accurate prenatal diagnosis is of utmost importance [8, 9, 10].
DDSH is a lethal autosomal recessive form of dwarfism with characteristic anisospondylic micromelia. Clinical features of DDSH include dwarfism, short and bowed limbs, flat facial features, anisospondyly, and encephalocele. The neonatal lethal condition DDSH is caused by functional null mutations to HSPG2, which completely prevent perlecan secretion into the extrcellular matrix [11].
The HSPG2 gene is an important, highly conserved gene whose expression affects many developmental processes, including the formation of the heart and brain systems, together with cartilage, bone marrow, and skeletal muscle [12]. A large gene encompassing 97 exons, it maps to chromosome 1p36.1 [11]. HSPG2 encodes the heparin sulfate proteoglycan 2 protein, perlecan, a co-receptor for basic fibroblast growth factor. Perlecan is a major component of basement membranes and is present in many tissues, including cartilage [13]. A type of skeletal dysplasia, DDSH is typically caused by functional mutations in the gene HSPG2, specifically due to a functional deletion mutation of the perlecan gene. In Silverman-Handmaker syndrome, the mutant perlecan molecules were unstable and cannot secreted into the extracellular matrix, and was degraded to smaller fragments with in the cells. Therefore, DDSH is caused by a functional null mutation of HSPG2 [14, 15].
Fewer than 40 cases have been reported in the literature, and only four of these cases were detected antenatally [16].
This study was carried out with the approval of the ethics committee (number:
SZFTFY-20191108). A Chinese pregnant woman, G3P1A1, 32 years old, was admitted to
the prenatal diagnosis center for genetic counseling (16
Family history: The first fetus was diagnosed with short limb malformation at
the 17th week of gestation. According to the B-scan ultrasound examination report
of the fetus, the biparietal diameter was 4 cm, abdominal circumference was 12.5
cm, femur length was 1.7 cm (
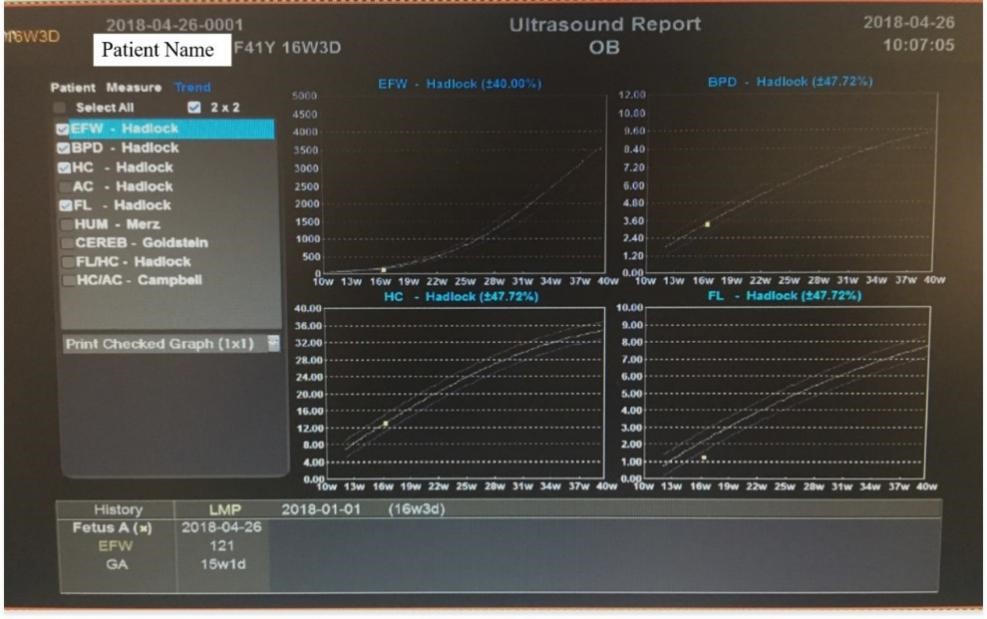 Fig. 1.
Fig. 1.The B-ultrasound report.
 Fig. 2.
Fig. 2.Ultrasound Atlas figures. (a) fetal humerus (14 w). (b) femur length (14 w). (c) fetal humerus (15 w). (d) biparietal diameter (14 w).
Amniotic fluid was extracted from the midline (about 1 cm right) of the lower abdomen of the pregnant woman (we determined the puncture point and collected amniotic fluid accurately under the guidance of abdominal ultrasound), then was detected by high through put sequencing. Subsequently, DNA was extracted by Gentra Puregene Blood Kit (Qiagen, Hilden, Germany). NanoDrop 2000 protein nucleic acid analyzer was used to detect DNA concentration and purity.
The venous blood of the couple was collected for high throughput sequencing. DNA was extracted by Gentra Puregene Blood Kit (Qiagen Company). NanoDrop 2000 protein nucleic acid analyzer was used to detect DNA concentration and purity. Positive mutation sites found by high throughput sequencing were validated using Sanger sequencing.
1
The identified positive mutation sites were validated in DNA extracted from parental blood and amniotic fluid by Sanger sequencing. The primer of HSPG2 gene was designed by the on-line primer design software. Subsequently, the primer was amplified by PCR and the target bands were identified by electrophoresis. The amplified products were sequenced by Sanger. The results of Sanger sequencing were analyzed by Mutation Surveyor V5.0.1 software (SoftGenetics, State College, PA, USA).
350 related genes were detected in this analysis. Therein, 5311 coding regions contained 835,566 base groups. The average coverage depth was 230+/–236x. Therein, 98.2% of the coverage area was greater than 10x and 96.8% of the coverage area was greater than 20x.
Two pathogenic mutations of HSPG2 gene were identified by high
throughput sequencing and validated by Sanger sequencing. Two heterozygous
mutations of HSPG2 gene were detected as c.6001dupC (p. R2001pfs*19) and
c.11207G
Next generation sequencing results were confirmed by Sanger sequencing.
Heterozygous mutation c.6001dupC (p. R2001pfs*19) and c.11207G
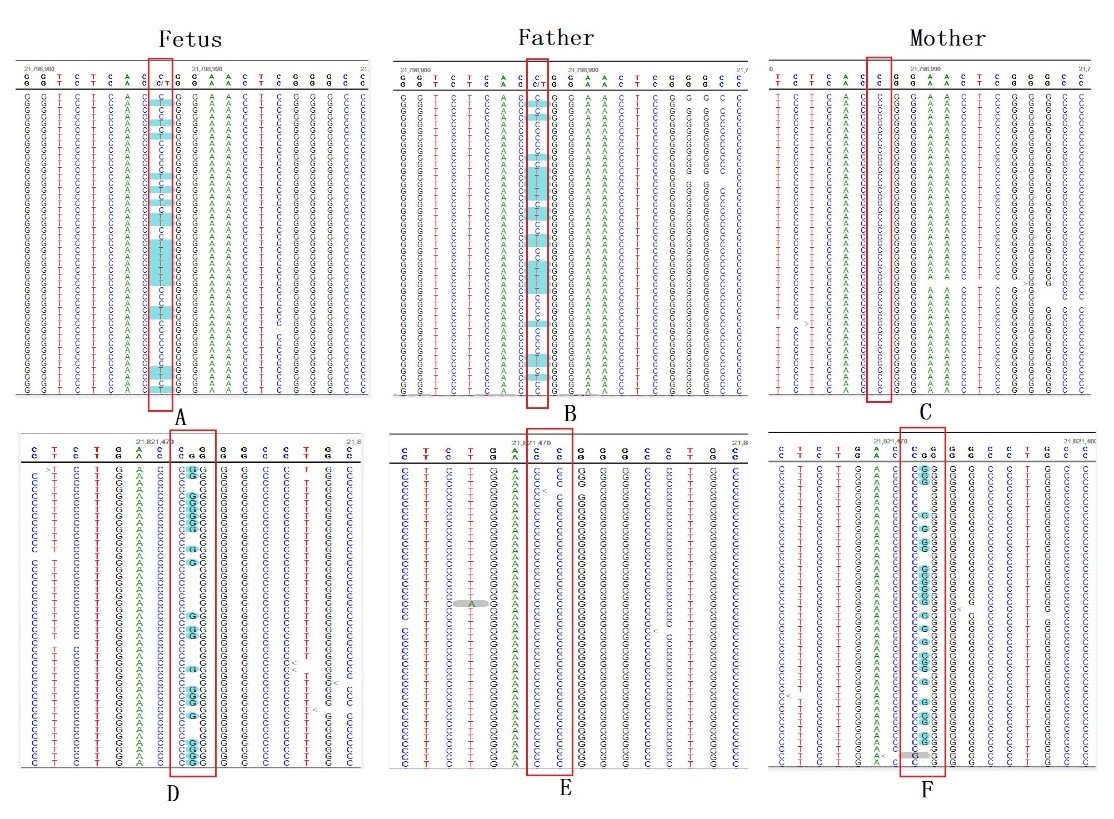 Fig. 3.
Fig. 3.Gene mutation map of fetus and parents. Mutation site: high
throughput sequencing site indicated that there were two mutation sites in fetal
HSPG2 gene: c.11207G
The pregnancy was terminated by induction of labor at 16 weeks as requested by the pregnant woman and her family. There was no autopsy (and therefore, no relevant pictures) due to familial disagreement.
Fetal short limb deformity is one of the most common birth defects in clinic, along with osteogenes is dysplasia, cartilage dysplasia, fatal dwarf, and short rib-polydactyly syndrome [17, 18]. The common pathogenic genes of fetal short limb deformity have been identified. For example, the pathogenic genes of achondroplasia, fatal dwarf and osteogenetic dysplasia are FGFR3 and COL1A1/COL1A12 [19, 20, 21, 22, 23, 24]. DDSH is generally caused by functional null mutations in the gene, HSPG2. The clinical manifestations are short limb dwarfs of newborns and may be accompanied by mental disorders [25] (PMID4059934, 3605216, 4953364, 2290482, 25666757). In this study, the whole exon of fetal gene was sequenced in a case of short limb malformation diagnosed by ultrasound, and the prenatal accurate gene diagnosis was completed. The parents did not show any clinical manifestations of the disease. These results indicate that the fetus short limb malformation may be caused by autosomal recessive inheritance.
The two identified variants of HSPG2 gene, c.6001dupC (p. R2001pfs*19)
and c.11207G
This is the first time these two mutations of HSPG2 genes are reported. The c.6001dupC (p. R2001pfs*1 mutation into a shift mutation) predicts a stop codon, which would result in a truncated version of the protein. The region where the p. R373Q mutation is located is an important component of the protein. The amino acid sequences of different species around this area are highly conserved. Computer-aided analysis predicts that the structure/function of the protein is likely to be affected.
In conclusion, novelmutations c.6001dupC (p. R2001pfs*19) and c.11207G
YXW designed the research study and performed the research. HW provided medical records and geneticsequencing for children and their parents. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of the hospital (approval number: 20180422).
The authors would like to thank the staff involved in the treatment of the patient, including Hui Wang, Xiaoyang Yang, Xian Chen et al. at the cooperating Departments at Medical Heredity Center, Shenzhen Maternal and Child Health Hospital, Southern Medical University. We wish to acknowledge the support of pregnant women and their families in helping us complete prenatal diagnosis and family follow-up.
This research received no external funding.
The authors declare no conflict of interest.