
- Academic Editor
Background: Trastuzumab (Herceptin®) is currently the main treatment option for breast cancer patients that overexpress the human epidermal growth factor receptor 2 (HER2). This antibody binds specifically to HER2, blocks cancer cell growth, and promotes effective cell death. In the present study, we sought to develop a robust and efficient process for the development of a stable Chinese hamster ovary (CHO) cell line with high trastuzumab expression and production. Methods: We adapted a process that combines transposon system-based vector construction, suspension cell culture, and a high selection process. The latter, involved enhanced green fluorescent protein (eGFP) expression, fluorescence-activated cell sorting (FACS), and semi-solid methylcellulose media. Results: The construction of trastuzumab as a humanized monoclonal antibody was achieved by subcloning the synthesized light and heavy chain sequences into a suitable piggyBac expression vector. The optimized piggyBac vector used for the expression of trastuzumab in CHO cells resulted in the production of trastuzumab and reached 4.24 g/L in the T1A7 clone after a 7-day batch culture. The T1A7 clone was selected after screening over 1500 clones. Conclusions: The current simple workflow ensures strict monoclonality and relatively high production of trastuzumab. This workflow could potentially be implemented in Research and Development (R&D) laboratories, including in developing countries for the production of recombinant monoclonal antibodies in a cost-effective manner.
Trastuzumab (Herceptin®), a humanized immunoglobulin G1 (IgG1) that targets the human epidermal growth factor receptor (HER2), is one of the top six selling monoclonal antibodies (mAbs) worldwide. It was the first monoclonal antibody (mAb) to be approved by the Food and Drug Administration (FDA) for the treatment of early breast cancer, advanced breast cancer, and gastric cancer, where HER2, a transmembrane protein encoded by the HER2/neu, is overexpressed [1, 2, 3, 4, 5, 6]. Unfortunately, access to trastuzumab has been hampered by its high cost. Hence, the development of trastuzumab biosimilars has been stimulated by the expiration of the European Union (EU) patent on trastuzumab. Thus, to facilitate the availability and accessibility of this expensive therapeutic class, the development of biosimilars is predicted to be between 20% and 30% less expensive than the reference molecules [7].
In the biopharma industry, expressing therapeutic mAbs that are high quality and titters in an economically viable process is vital. Achieving a robust and controllable process for antibody production is a highly complex and sensitive pathway that requires careful consideration of many factors and parameters, which includes choosing the host, gene amplification strategy, expression vector types, and clone screening techniques [8]. Nowadays, 95% of the therapeutic mAbs are produced in mammalian cell lines [9]. Chinese hamster ovary (CHO) cells are currently the most widely used host cells in medical Research and Development (R&D) laboratories and the biopharmaceutical industry in producing mAb-based biologics. They can ensure the correct protein assembly, folding, and sophisticated post-translational modifications (i.e., glycosylation, phosphorylation), the exact same as human cells [10, 11]. CHO cells are also of interest to pharmaceutical companies owing to their growth rate in serum-free media, high yield (up to 10 g/L), scalability, manufacturability, and utility in conjunction with selective markers, such as specific antibiotics, methotrexate (MTX), or glutamine synthetase (GS) [12, 13].
While the high production capacity of a recombinant protein from a selected cell
clone is economically important, consistency and stability over generations of
cell cultures and fed-batch process cultures are critical for biopharmaceutical
companies. Therefore, the choice of expression vector used to transfect the cell
lines is a key factor in maintaining high productivity. Several studies have
shown that random integration of the transgene into the genome of cells is the
main factor limiting the stability of protein expression [14, 15, 16, 17]. Unlike
conventional transfection methods, which typically use vector-based random
integration of foreign genes into chromosomes, site-specific recombination
strategies offer effective alternatives for developing productive and stable
clones [18]. Thus, this represents an alternative approach to obtaining constant
protein expression of transgenes in transposon systems [19, 20, 21]. The PiggyBac
transposon system (PB system) is one of several transposons that has been
engineered to be functional in mammalian cells. The system combines a helper
plasmid, which encodes the transposase gene (PBase), and a donor vector featuring
the 5
In the present study, the PiggyBac system was used for the purpose of mAb expression. A helper vector and a donor containing trastuzumab heavy- and light-chain sequences were synthesized. These sequences consisted of optimized signal sequences to ensure the proper antibody folding and post-translational processing. Using puromycin as a selection pressure, the expression vector was stably expressed in the CHO-S cell line. Thereafter, the ability of the CHO-S cells to produce the target antibody was assessed. By outlining a simple workflow, the main objective of this work was to establish the first Moroccan biotechnology platform to produce recombinant proteins, including mAbs. This could strongly encourage the Moroccan pharmaceutical industry to adopt such platforms and produce biologics, which are currently imported. This will result in long-term savings on costs in the Moroccan healthcare system, thereby improving their affordability and accessibility to Moroccan patients.
All cell lines were validated by Short Tandem Repeat (STR) profiling; Mycoplasma testing was performed in all cell lines every two weeks using the MycoSEQ™ Mycoplasma Detection Kit (Thermo Fisher Scientific, Waltham, MA, USA).
The trastuzumab expression cassette was designed in-house. Indeed, the trastuzumab heavy-chain (Tras-HC) and light-chain (Tras-LC) sequences were obtained from the Protein Data Bank (PDB) (1N8Z) and Drug Bank (DB00072) databases. Cytomegalovirus (CMV) promoter was selected to drive Tras-HC and Tras-LC expression. Then, the dual reporter gene for enhanced green fluorescent protein (eGFP) and puromycin was inserted under the control of the CMV promoter, using HindIII and SpeI restriction sites and terminated by BGH pA. The gene encoding the IL-2 secretory signal sequence was inserted upstream of Tras-HC and Tras-LC and three different poly A elements were inserted upstream of the CMV promoter. Then, the expression cassette was cloned into the donor vector (Piggy-Tras), and while the Piggy-Tras construct was generated by commercial synthesis (Vector Builder, NJ, USA).
Next, Escherichia coli DH5-alpha cells were transformed using the Piggy-Tras and Piggy-Trans plasmids. Sufficient plasmids were prepared for transfection. Following transformation, clones corresponding to each of the vectors were randomly selected for purification. Recombinant plasmids were purified from bacterial cells using the PureLink HiPure Plasmid Filter Maxiprep Kit (Thermo Fisher Scientific, Waltham, MA, USA).
The purified Piggy-Tras were amplified by PCR for Tras-HC and Tras-LC using
primers (forward-HC: 5
The suspension Freestyle CHO-S cells (Thermo Fisher Scientific, Waltham, MA,
USA) were maintained in FreeStyle™ CHO expression medium (Thermo
Fisher Scientific, Waltham, MA, USA). The chemically defined, protein-free
medium was supplemented with 8 mM L-glutamine (Invitrogen, Carlsbad, CA, USA), 2
Mm Pen-Strep (Invitrogen, Carlsbad, CA, USA), and an anti-clumping agent
(Invitrogen, Carlsbad, CA, USA). The cells were kept in a humidified incubator on
a shaker at 37 °C with 8% CO
Lipofectamine 3000 reagent (Life Technologies, Carlsbad, CA, USA) was used to
co-transfect the FreeStyle CHO-S cells in triplicate. Twenty-four hours prior to
transfection, 30 mL of FreeStyle™ CHO expression medium containing
6
Before transfection, an enhanced stringency selection of the CHO-S cells was performed using a killing curve, which used puromycin concentrations ranging from 20 to 100 µg/mL (Sigma-Aldrich, St. Louis, MO, USA). Following, 48 hours of transfection, the cell media were changed and supplemented with the optimal puromycin concentration. Thereafter, the selective medium was replaced every three days for a month to obtain a stable pool, the stability of which was evaluated by fluorescence microscopy and sodium dodecyl polyacrylamide gel electrophoresis (SDS-PAGE).
The level of mAb expression in the cell culture supernatants (plates and shake
flasks) was determined by indirect ELISA. The concentration of the secreted mAb
was determined using the Herceptin Trastuzumab/Anti-HER2 Humanized IgG ELISA Kit
(Alpha Diagnostics International, San Antonio, TX, USA). Briefly, 8-well strips
were used, which were pre-coated with HER2 antigen and post-coated with
stabilizers. Standards and culture supernatants were diluted, loaded into each
well, and treated with anti-human IgG-horseradish peroxidase (HRP) conjugate. TMB
(tetramethylbenzidine) (Sigma-Aldrich, St. Louis, MO, USA) and HRP substrate were
added to the plates. After incubating for approximately 10–20 min at room
temperature in the dark, sulfuric acid (H
Fluorescence microscopy was used to monitor the stability of the cell pool in the shake flask during a one-month culture without selection pressure via the eGFP expression. The cells were washed twice with 1 mL of PBS, and fixed in 1 mL of ice-cold 100% methanol for 10 min before being washed again with 1 mL of PBS. The coverslips were mounted in ProLong Diamond Antifade Mountant and DAPI was applied (Life Sciences, Cambridge, MA, USA). The images were acquired using an EVOS® FL Cell Imaging microscope.
Since the donor vector was harboring eGFP, the encoding sequence transfection rates in the cells were determined 48 hours post-transfection. The cells were cultivated for two days at 37 °C before being resuspended in PBS. The FACSCalibur™ system (BD Biosciences, San Jose, CA, USA) was used to estimate the green fluorescence intensity of various clones. Green fluorescence at 525 nm was detected through an FL1 set at a PMT voltage of 400, with a logarithmic gain. Ten thousand cells were analyzed for each sample.
The methylcellulose-based semi-solid medium was prepared in-house using a
minimum essential medium (MEM) 10X (Gibco, Waltham, MA, USA), 2% methylcellulose
4000 pentose (Sigma-Aldrich, St. Louis, MO, USA), 1% non-essential amino acids
(Gibco, Waltham, MA, USA), 14 mM NaHCO
The high clone producer T1A7 was cultured in triplicate in a batch culture mode,
in a 125 mL Erlenmeyer flask containing 30 mL of FreeStyle™ CHO
expression medium (Thermo Fisher, Waltham, MA, USA) supplemented with 8 mM
glutamine (Invitrogen, Carlsbad, CA, USA), and 1% anti-clumping agent
(Invitrogen, Carlsbad, CA, USA). Flasks were inoculated with 1
After 7 days of batch culture, clarification was carried out on the T1A7 clone
by centrifuging the cells at 5000
Purified mAb produced from the T1A7 clone was analyzed by SDS-PAGE under reducing conditions. Samples were heated at 95 °C for 5 min and loaded into the gel wells. As a negative control, 10 µL of non-transfected CHO-S cells supernatant was loaded alongside 10 µL of the three purified samples. Coomassie blue staining was performed to visualize the resulting protein bands.
The protein aggregates were analyzed using an Agilent HPLC system (1260 Infinity
II series). For that purpose, an Agilent AdvanceBio size exclusion chromatography
(SEC) column was used with the following characteristics: 7.8
Statistical analysis was carried out using GraphPad Prism software (version
9.2.0 (283), GraphPad Software, Inc., San Diego, CA, USA), with two-tailed p-values below 0.05
being considered statistically significant, and a 95% confidence interval
(*p
The generated Piggy-Trans and Piggy-Tras vectors were obtained from Vector
Builder, Englewood Cliffs, NJ, USA. The expression cassette designed to express
the full-length trastuzumab (Tras-HC sequence encoding 451 amino acids and the
Tras-LC encoding 215 amino acids) was inserted between the 5
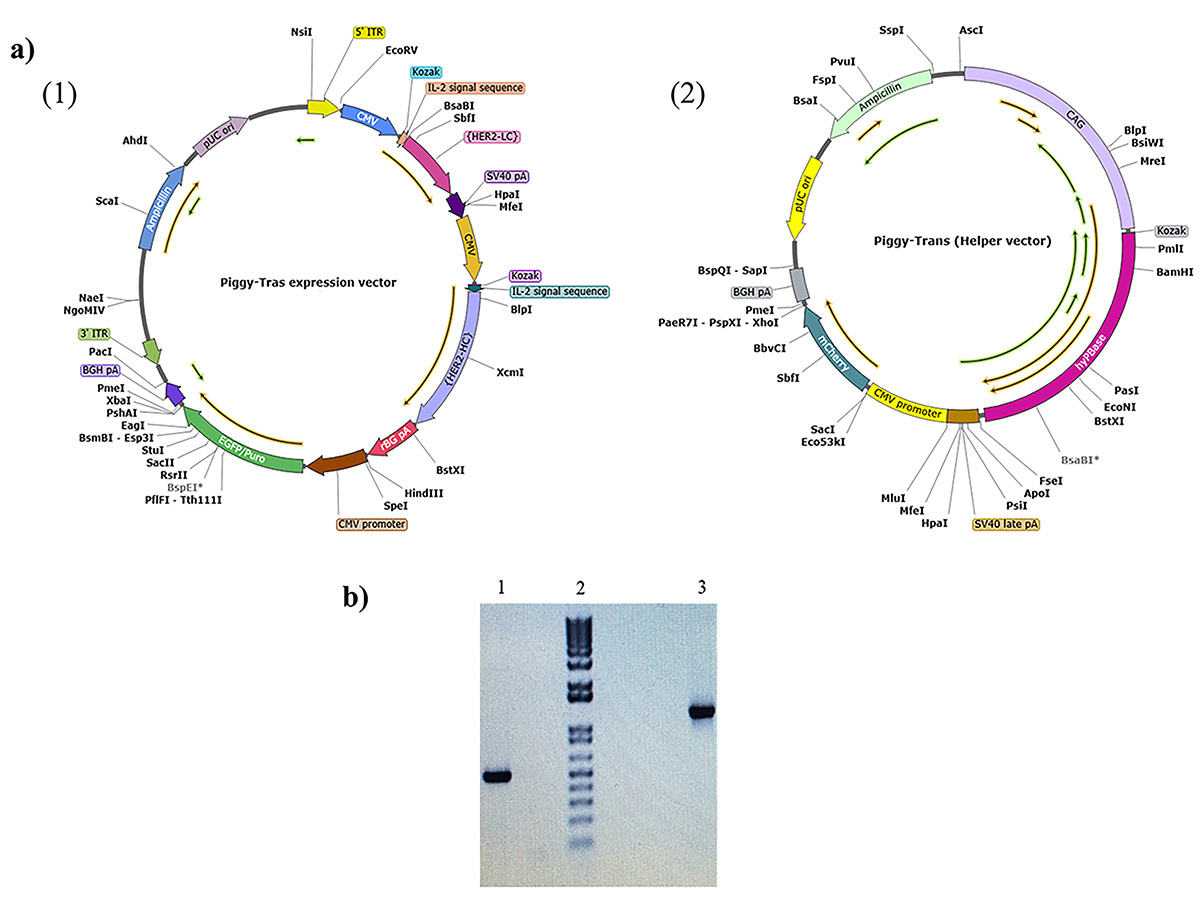 Fig. 1.
Fig. 1.Representation of the PiggyBac vector maps for
Piggy-Tras and Piggy-Trans and agarose gel illustrating Tras-HC and Tras-LC
insertion. (a) Piggy-Tras and Piggy-Trans maps. (1) Piggy-Tras vector containing
the expression cassette encoding the full-length trastuzumab (Tras-HC and
Tras-LC) under the control of the cytomegalovirus promoter (CMV). It also harbors
the pressure selection genes (puromycin and ampicillin) and enhanced green
fluorescent protein (eGFP). (2) Piggy-Tras encoding the transposase under the
control of the enhancer-chicken
To generate a stable CHO-S cell line, a puromycin kill curve representing a dose-response experiment was performed to determine the minimum concentration of puromycin that can kill all the cells during a specific period. Twenty-four hours after culturing the non-transfected CHO-S cells, the growth medium was replaced with a selection medium that had been supplemented with a range of puromycin concentrations (20 µg/mL–100 µg/mL). A concentration of 50 µg/mL was determined as the minimum concentration that would kill all the cells within 9 days (Fig. 2).
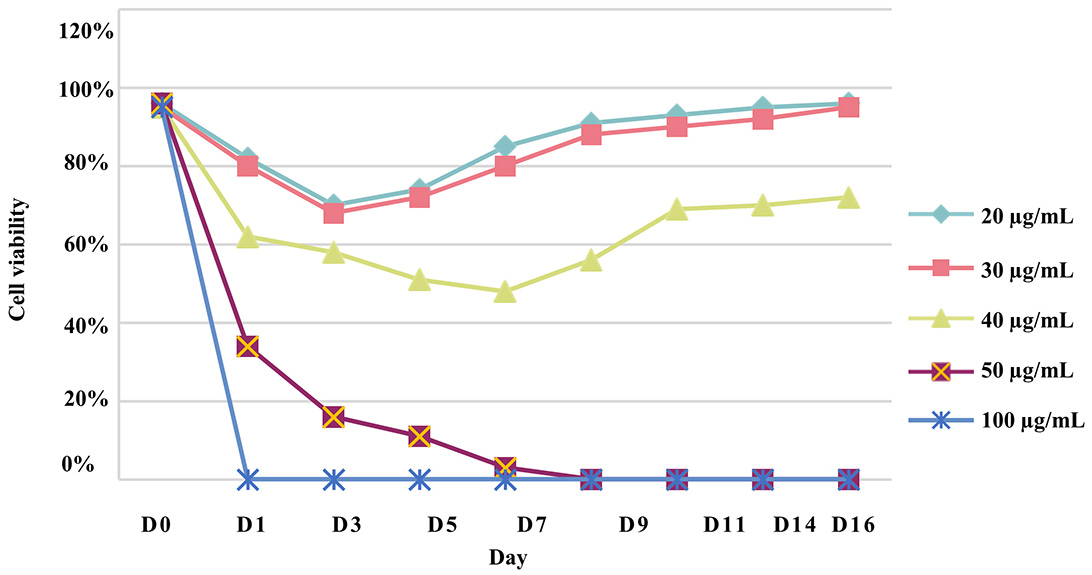 Fig. 2.
Fig. 2.Chinese hamster ovary (CHO)-S cell puromycin kill curve. To determine the optimal concentration for selecting transfected cells, a puromycin concentration range of 20 µg/mL to 100 µg/mL was used on CHO-S cells for two weeks and performed in triplicate.
The impact of the Piggy-Tras:Piggy-Trans ratio on the generation of a stable CHO-S pool was also investigated. For that purpose, cells were co-transfected in triplicate with three Piggy-Tras/Piggy-Trans ratios (1:3, 1:5, and 1:10) to determine the optimal conditions for efficient insertion of the gene of interest (GOI) into the host cell genome, and to generate an efficient cell pool with a high mAb expression and production [29, 30]. The transfection supernatants of the three ratios were collected and the secreted antibodies were evaluated using the human IgG ELISA kit. mAb quantification showed that a 1:3 transfection ratio of Piggy-Tras/Piggy-Trans was optimal in CHO cells to obtain higher titers of secreted antibodies (5.1 µg/mL), compared to 0.58 µg/mL for the 1:5 ratio and 0.38 µg/mL for the 1:10 ratio (Fig. 3). Subsequently, the cells from the 1:3 ratio pool were maintained in the selection medium (50 µg/mL of puromycin) for one month. The cell viability was constantly monitored during the 1-month generation of stable cells. During the first days of culturing under selective conditions, the proportion of viable cells was very low (12%) compared to the non-transfected cells (88%). Gradually, the cell viability of the transfected cells improved and reached 98% on day 31 (Fig. 4a). Fluorescence microscope showed that most of the CHO-S cells in the stable pool expressed GFP (Fig. 4b(B)), while non-transfected cells (Fig. 4b(A)) show no expression of GFP. In addition, SDS-PAGE, under reducing conditions, showed that the migration of the cell pool supernatant (in triplicate) was characteristic of an antibody, with the HC and LC bands observed at the expected sizes of 50 kDa and 25 kDa, respectively (Fig. 4b(C)).
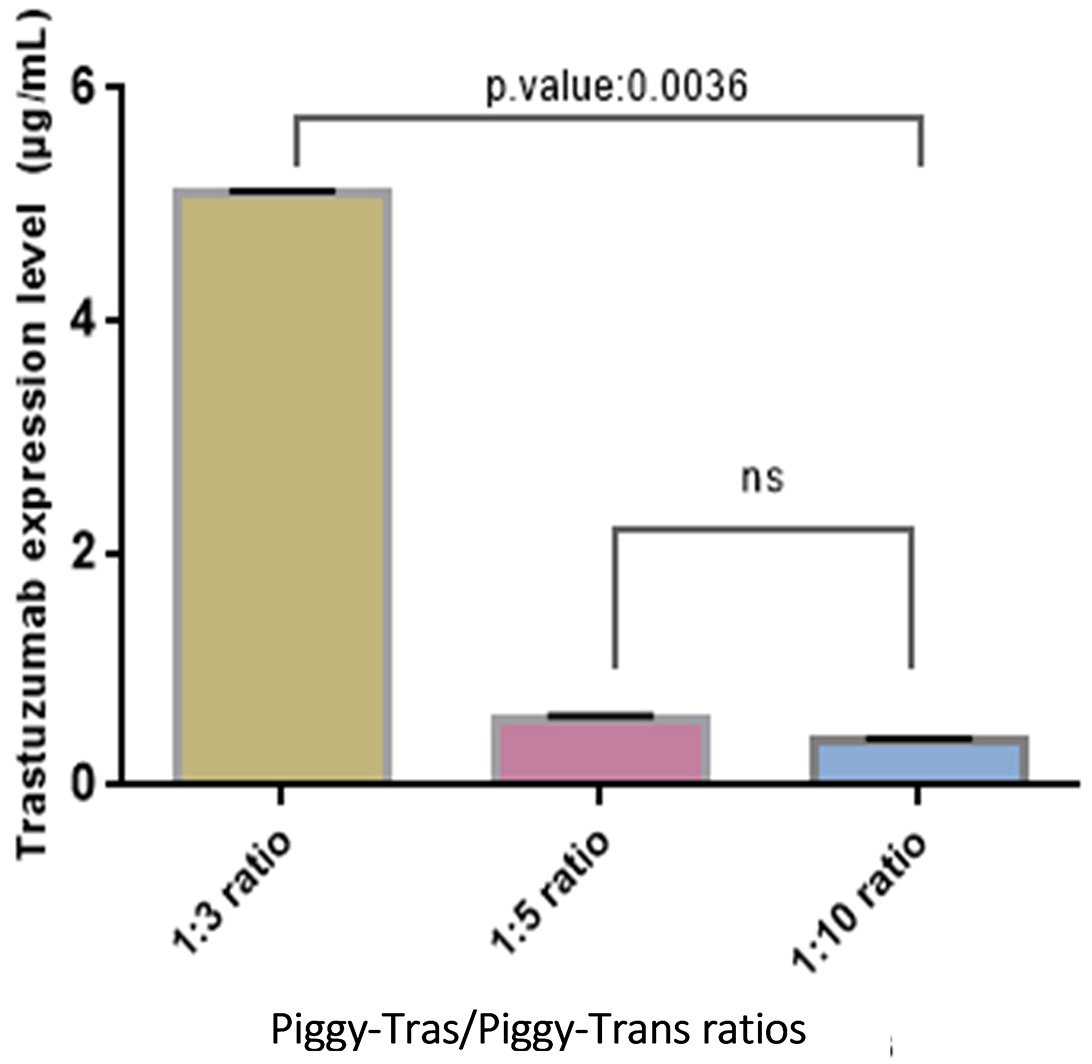 Fig. 3.
Fig. 3.Impact of Piggy-Tras/Piggy-Trans ratio on antibody expression. Three pools of CHO-S cells were generated (in triplicate) at three different ratios: 1:3, 1:5, and 1:10. Following 4 days of co-transfection, mAb titers were measured using a human IgG enzyme-linked immunosorbent assay (ELISA) kit. Error bars represent the average SD from triplicate measurements. ANOVA (Kruskal–Wallis test) was used to detect statistically none significant (ns) and significant differences between the generated pools.
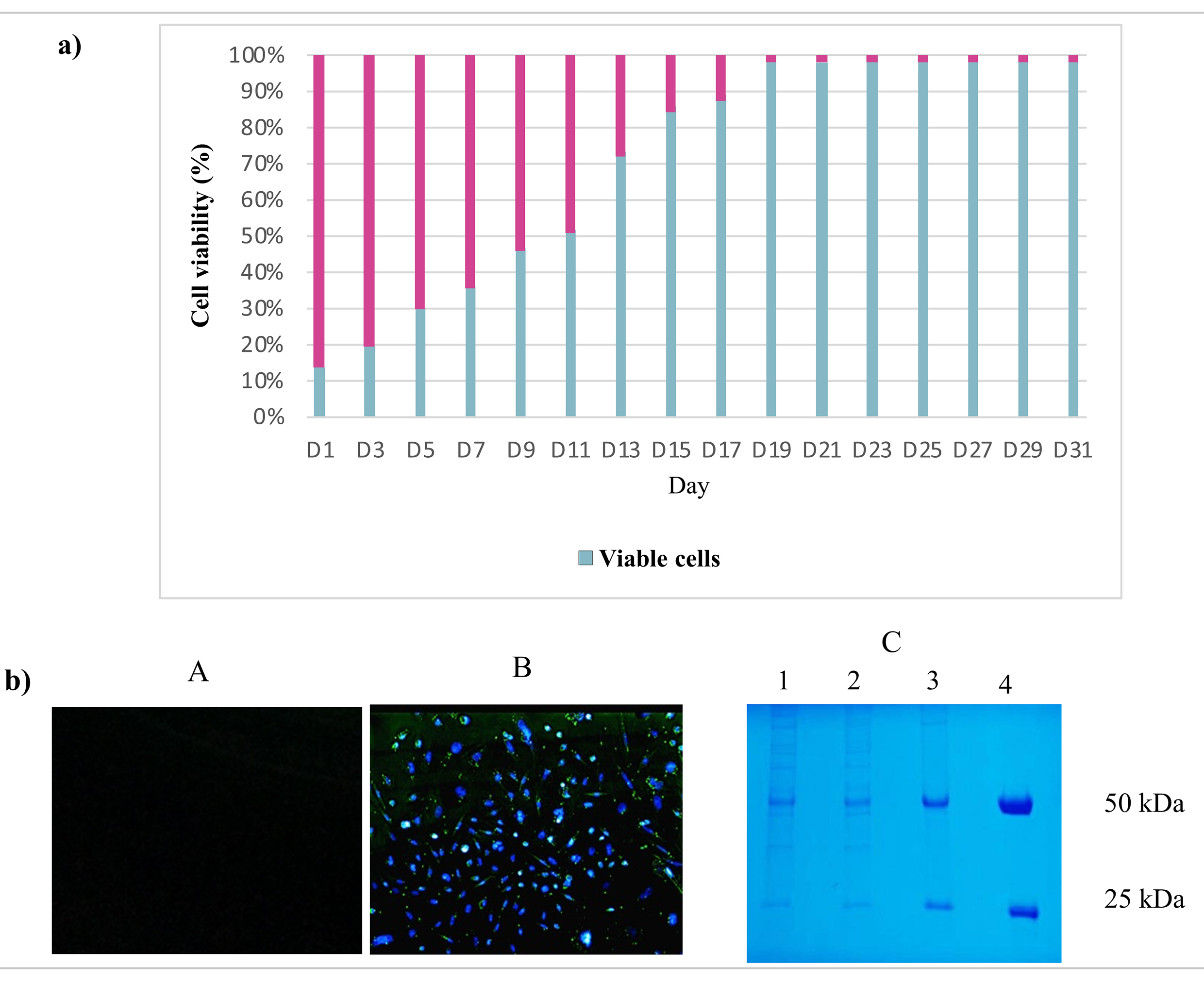 Fig. 4.
Fig. 4.Fluorescence microscopy and sodium dodecyl polyacrylamide gel electrophoresis (SDS-PAGE) evaluation of stable pool generation. (a) Stable cell pool generation: During one month of cell culture under puromycin selection, the CHO-S cell viability was measured. Indeed, the CHO-S cell viability was low (12%) during the first days but increased to 98% by day 23. (b) Fluorescence microscopy and SDS-PAGE evaluation of the stable pool. (A) The supernatant of non-transfected CHO-S cells was visualized by fluorescence microscopy and used as a negative control. (B) The CHO-S stable pool shows the expression of eGFP protein, which is proportional to the expression of the mAb. (C) SDS-PAGE analysis following Coomassie blue staining: migration of the CHO-S stable cell pool supernatants (in triplicate: lane 1, 2, 3) in SDS-PAGE (10%) under reducing conditions showing the expected bands, 50 kDa for Tras-HC and 25 kDa for Tras-LC. Lane 4 refers to the positive control: commercial Herceptin® (Roche, Basel, Switzerland).
The strategy of isolating stable CHO-S clones was based on the combination of
fluorescence-activated cell sorting (FACS) followed by methylcellulose semi-solid
media. The FACS Calibur system was used to purify the stable cell pool based on
the eGFP expression. The eGFP fluorescence was detected in the FL1 channel, and
the population of interest was gated to sort the sterile selected CHO-S
population (Fig. 5a). The cells were collected for subsequent culturing in 2%
MCM. The medium formulation was optimized in-house and included basal 10
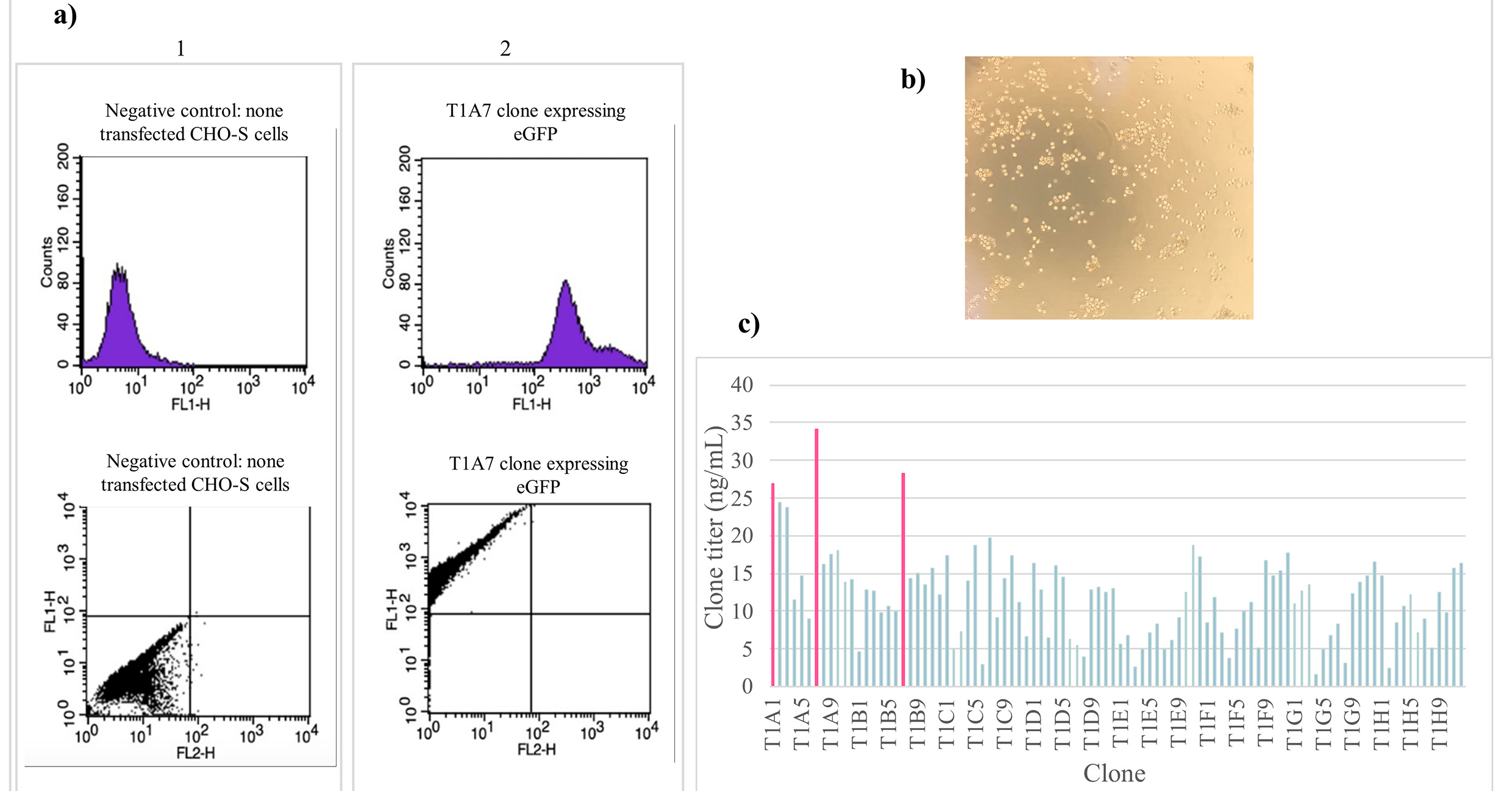 Fig. 5.
Fig. 5.CHO-S cell sorting and clone generation. (a) Cell
sorting analysis was performed on the stable pool of CHO-S cells. GFP
fluorescence was detected in the FL1 channel and the population of interest with
high eGFP expression was gated for sterile sorting. (b) Following the cell
sorting, the cells were cultured in optimized 2% MCM, the figure shows the
clones generated after 12 days of incubation in a humidified 8% CO
The T1A7 clone was chosen for the triplicate batch culture investigation.
Indeed, 30 mL of Freestyle CHO expression medium was inoculated with the clones
seeded at a density of 1
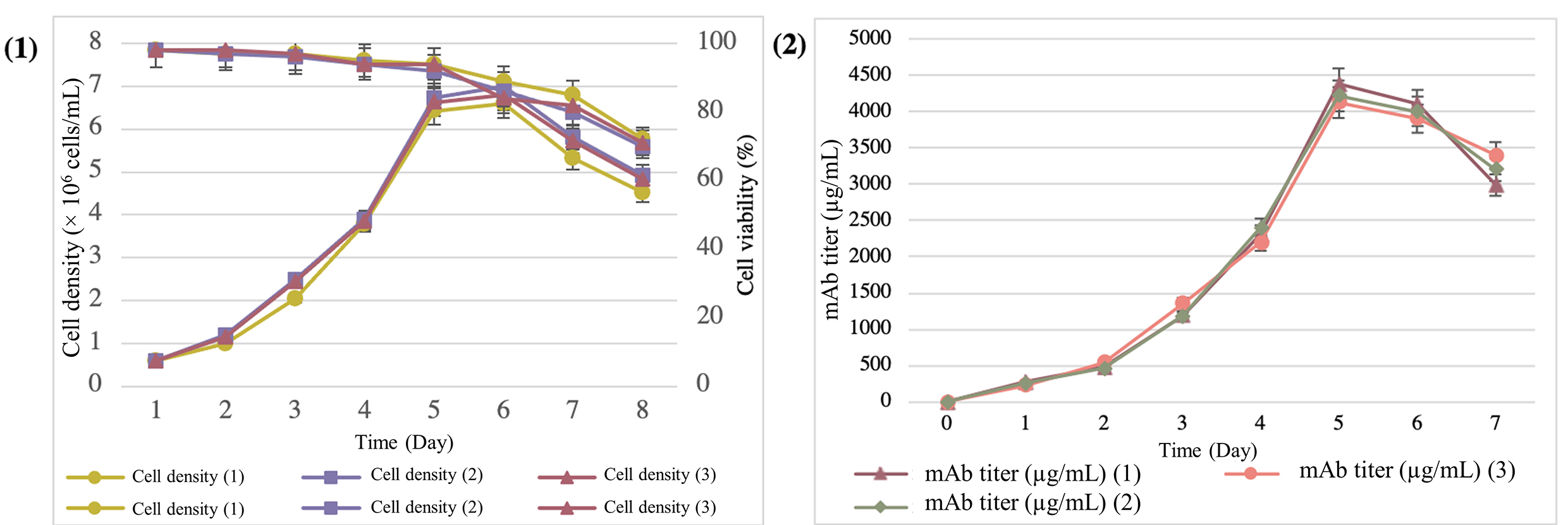 Fig. 6.
Fig. 6.Expression and quantification of mAbs from the supernatant of the T1A7 clone during the 7-day batch-culture mode. (1) Cell density and viability during the batch-culture mode. (2) Determination of the trastuzumab titer by the ELISA Herceptin Kit.
The purity and size of Herceptin® (Roche, Basel, Switzerland) and the mAbs secreted by our T1A7 clones were analyzed by SDS-PAGE and SEC-HPLC. Indeed, the molecular size of Herceptin® and the purified mAbs secreted from the T1A7 clones were also analyzed by SDS-PAGE, under reducing conditions, and showed that the protein bands of both molecules matched, while there was also an absence of any other low or high molecular weight protein bands (Fig. 7b).
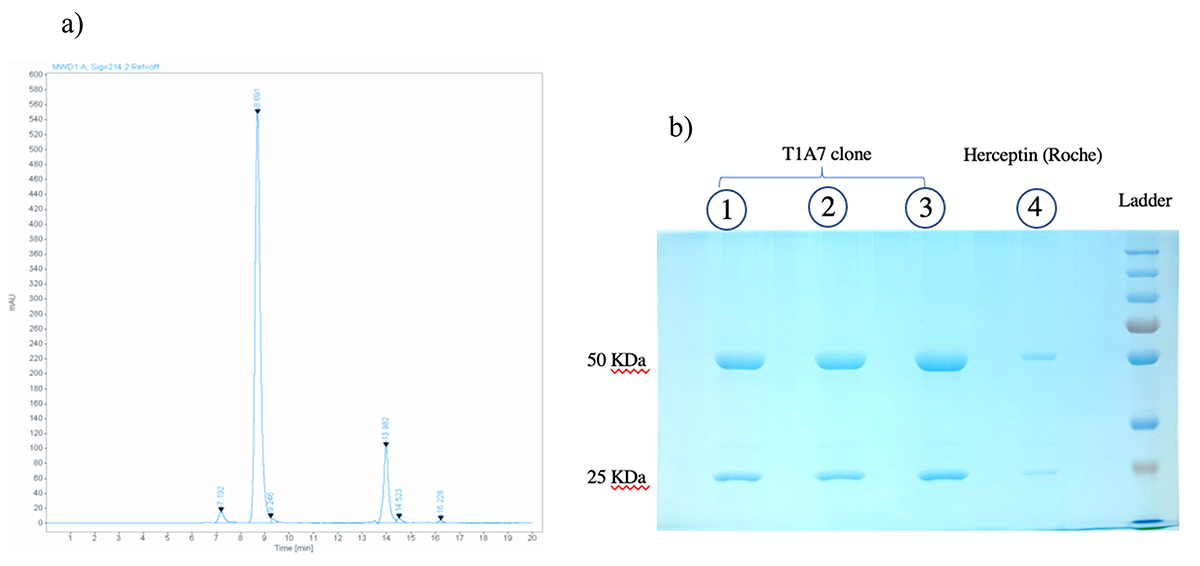 Fig. 7.
Fig. 7.Size and aggregate analyses. (a) SEC chromatogram obtained from SEC-HPLC showed a main peak with a mass of 165,000 g/mol that was eluted at 8.691 min and a mass of 3000 g/mol that was eluted at 13.982 min. (b) SDS-PAGE analysis of the purified mAbs from the T1A7 clone following Coomassie blue staining: after protein A purification of mAbs from the T1A7 clone supernatant, mAb migration (in triplicate: lane 1, 2, 3) in SDS-PAGE (10%) under reducing conditions showed the expected bands, 50 kDa for Tras-HC and 25 kDa for Tras-LC. Lane 4 refers to the positive control: commercial Herceptin® (Roche).
According to the calibration curve, the purity of the mAbs secreted from the T1A7 clone using SEC-HPLC was 95%, with a retention time of 8.691 min for the main peak, and a mass of 165,000 g/mol. In addition, 3% high molecular weight (HMW) aggregates were present (Fig. 7a), without any low molecular weight (LMW) aggregates.
A renewed focus on rapid, cost-effective cell line development and predictable product quality attributes is required in the biosimilar market. Traditional stable cell line development involves selective pressure for clone isolation and random integration of the gene of interest (GOI) into the host cell genome, to potentially provide heterogeneity and unstable expression. Therefore, high and sustained antibody expression are certainly two key attributes in the selection of a CHO cell line development strategy. To this end, several studies have recently been conducted on the use of PiggyBac technology in CHO cells for the production of proteins of interest [31, 32, 33, 34].
In the present study, we used PiggyBac technology to develop, for the first time in Morocco, a cell line that produces trastuzumab antibodies. This technology specifically targets stable regions of the genome with highly expressed genes. It is based on mobile genetic elements that can be transposed between genomic sites by a “cut and paste” mechanism, allowing the GOI to integrate into the tetranucleotide TTAA chromosomal sites distributed throughout the CHO cell genome [20, 22]. To achieve high levels of antibody expression, vector design requires the selection of promoters, signal peptides, and codon optimization elements. Therefore, the Piggy-Tras possessed the strong CMV promoter used to drive the expression of Tras-HC and Tras-LC. To increase the export of the transcribed messenger RNA (mRNA) from the nucleus to the cytoplasm, an IL-2 intron sequence was inserted upstream of the Tras-HC and Tras-LC. The IL-2 intron sequence is also responsible for transporting the expressed antibody to the endoplasmic reticulum for folding, assembly, and post-translational modification before being secreted into the extracellular environment. The BGH pA and SV40 late polyadenylation signal sequence was also inserted to promote mRNA polyadenylation and splicing and to maximize mRNA levels. A consensus Kozak sequence was inserted upstream of the first translation initiation codon to enhance the initiation of translation (Fig. 1a) [35]. The transposase generated from the Piggy-Trans (Fig. 1a(2)) would recognize the two ITRs on the Piggy-Tras and insert the flanked region into the host genome. Integration typically occurs at sites on the host chromosome that contain the TTAA sequences that are duplicated on both flanks of the integrated cassette [36].
In addition to PB technology, the major goal of this study was to establish a stable CHO-S cell line capable of maintaining expression levels over time. To this end, a stable pool of cells was generated under puromycin pressure selection for one month. Despite the advent of highly efficient transient transfection protocols, generating stable mammalian cell lines remains critical for further investigating mAb functions and structures. Protein production using stable cell lines can be easily scaled up to achieve high product yields [32, 37, 38]. Fluorescence microscopy and Coomassie gels were used to verify the stability of the generated cell pool. Several studies have demonstrated a tight expression relationship between eGFP and the gene of interest (GOI), which is achieved when the GOI is cloned into a bicistronic vector together with a fluorescent protein [39]. As shown in Fig. 4b(B), the CHO-S cell pool expressed GFP on the 31st day, and the migration of the cell pool supernatant in SDS-PAGE, under reducing conditions, revealed the characteristic antibody bands at the expected sizes—50 kDa for HC and 25 kDa for LC (Fig. 4b(C)).
The isolation and screening strategy for stable CHO-S clones was based on the combination of two sequential steps: fluorescence-activated cell sorting (FACS) followed by a semi-solid methylcellulose medium. FACSCalibur was used to purify the stable cell pool based on eGFP expression (Fig. 5a), while our optimized 2% MCM was used to successfully expand the FACS-purified CHO-S population. Indeed, MCM has the advantage of restricting the movement of individual cells and preventing them from re-aggregating. At the same time, it is soft enough to support the growth of 3D-CHO cells in vitro [40].
Once the clones were generated, they were examined under the microscope to confirm that they successfully met the selection parameters (size, compactness, and distance between colonies). Amongst the 100 clones selected, T1A1, T1A7, and T1B7 clones showed the best titers with mAb concentrations of 26.87 ng/mL, 34.16 ng/mL, and 28.30 ng/mL, respectively (Fig. 5c). These results demonstrate that using the above sequential screening workflow, including cell population purification by FACS followed by our optimized MCM (2%) expansion, can provide high assurance of monoclonality.
The T1A7 clone, which produced the highest mAb titer, was selected and seeded
for 7 days in a batch culture. In terms of productivity, we observed an average
peak cell density of 6.79
Regarding protein size and aggregates, we noted the presence of 3% high molecular weight (HMW) aggregates without any low molecular weight (LMW) (Fig. 7a). In a related study, Joshi et al. [48] reported the presence of 2.20% HMWS of a biosimilar compared to 0.90% aggregates of the trastuzumab originator molecule. Cation exchange chromatography (CEX) could be applied to increase the mAb purity and eliminate mAb aggregates, especially following the optimization of the NaCl concentration and pH [49]. Indeed, protein aggregation is a common issue that can occur during various stages of bioprocessing, including cell culture, protein A purification, and formulation. Implementing appropriate strategies to control the development process and prevent aggregates would be of critical interest to minimize the risk of immunogenicity. Several studies have reported the need to explore strategies based on process optimization and understanding the aggregation mechanisms to prevent mAb aggregation [50, 51, 52, 53].
Ultimately, in addition to the simple and efficient cell line development described herein (Fig. 8), the T1A7 clone is a promising candidate for producing high yields of trastuzumab.
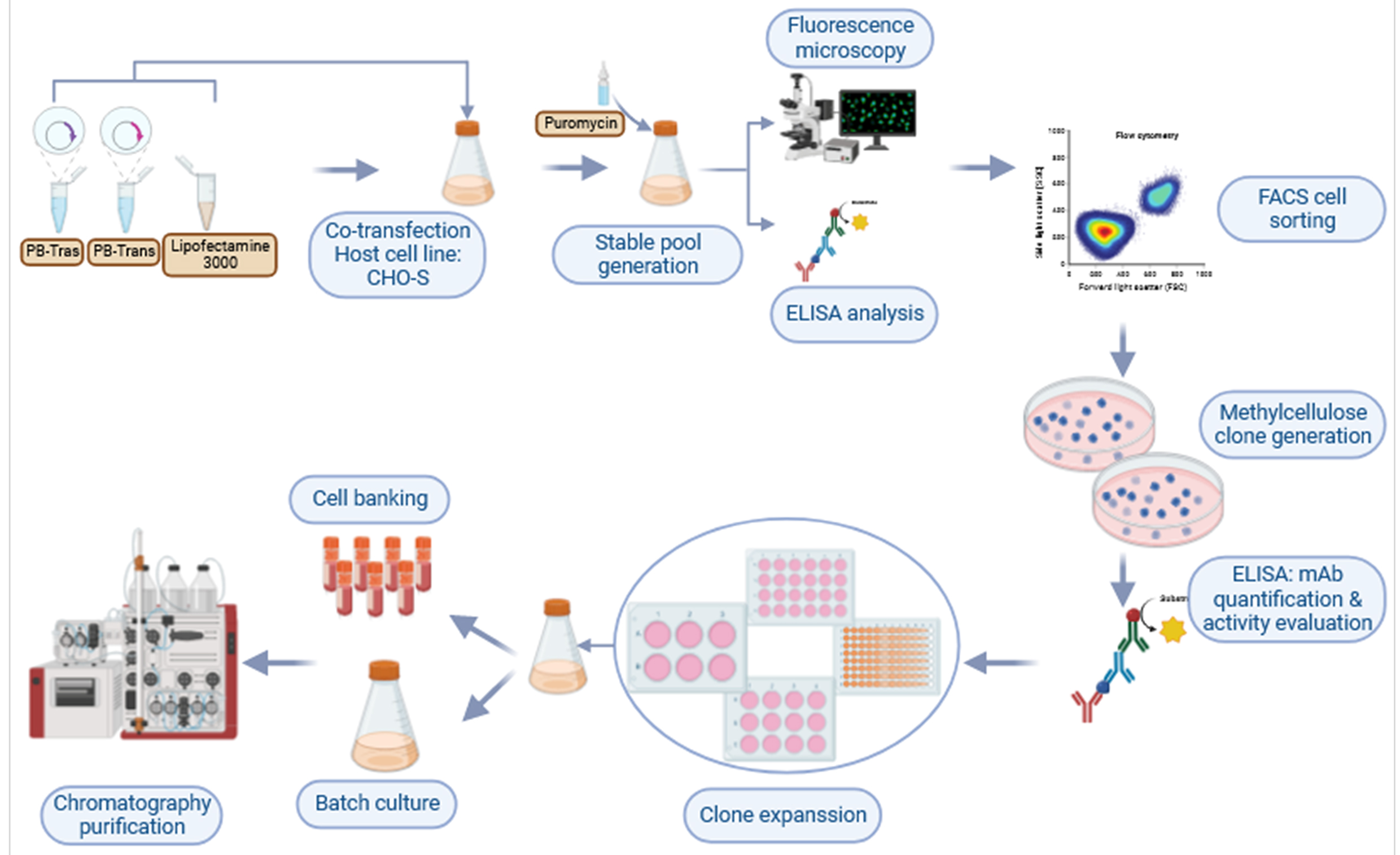 Fig. 8.
Fig. 8.Graphic representation showing the workflow used to develop a CHO cell line that produces high titers of trastuzumab.
The current study was the first attempt in Morocco to develop stable mammalian cells that produce recombinant mAbs. Our first established biotechnology platform allowed us to apply a simple strategy for the rapid development of CHO clones that produce good trastuzumab titers up to 4.24 g/L, which can be highly improved in a future pilot fed-batch process.
Our workflow combines PiggyBac-based vector technology, CHO suspension cell line, GFP expression monitoring, and dual FACS/MCM (2%) selection processes. It can be implemented in R&D laboratories, including in low- and middle-income countries (LMIC) for the development of cost-effective monoclonal antibodies and biosimilars with strict monoclonality assurance in a short and controlled timeframe (Fig. 8).
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
HAB conceptualized the study. HAB, HS, YC, and MYAF designed the study and secured funding. HB, NB and YA participated in designing & performing the experiments. HB wrote the original draft and final manuscript. HAB reviewed and edited the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript.
Not applicable.
The authors thank MAScIR Foundation and Mohammed VI Polytechnic University.
This work was funded by the Moroccan Foundation for Advanced Sciences, Innovation and Research (MAScIR) and the Moroccan MESRSF in the frame of the project PPR-C.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.