
- Academic Editor
Inflammation has been confirmed to exist in the tumor microenvironment, while the risk of cancer occurrence increases in cases of chronic inflammation. It is estimated that approximately 10% to 20% of cancers are associated with chronic infections and attendant inflammation. Bacteria, both pathogenic and commensal, viruses, and fungi actively participate in the development and maintenance of inflammation and tumor growth in humans. The exposome, which is a sum of human environmental exposures, such as industrial diet, consumed drugs, and toxins, affects the composition and function of the human microbiome, which could lead to dysbiosis and disorders in tissue homeostasis through different mechanisms, including the intensification of the immune response, activation and abnormal proliferation, and disruption to epithelial barrier integrity. Presently, science remains at the stage of revealing the complexity associated with the mechanisms involved in building relationships that cover the microbiome–inflammation–tumor, yet it is already known how important it is to care for microbial homeostasis of the organism.
The microbiome is now defined as an organ whose role is to supplement and
complement the proper functioning of the host organism. Considering this line of
reasoning, it should be assumed that multicellular organisms are no longer
independent, separate units but constitute the so-called holobionts—an organism
with its accompanying microbiota, which consists of viruses, bacteria, fungi, and
archaea. The components of the holobiont live in a state of dynamic equilibrium
and are subject to changes forced on them by the environment [1, 2]. The classic
definition of the human microbiome includes the microorganisms that inhabit it
and their structural elements, such as nucleic acids and metabolites. The vast
majority of the estimated 3.8
The intestinal microbiome in healthy people changes throughout life, meaning that the species composition of the microorganisms that colonize people differs between individuals. This also applies to the variability and diversity in the composition of the commensal microflora, which is a potential source of phenotypic variability in the development of the disease and therapeutic effectiveness. Thus, this variability has a huge impact on the carcinogenesis process. It also determines the therapeutic response, the characteristics of antitumor immunity, and the clinical response to immunotherapy [6].
The relationship between inflammation and carcinogenesis and the participation of microorganisms inhabiting the human body in these processes have been of interest to many scientists for over a century, with Virchow first showing the link between inflammation and cancer more than 150 years ago by observing leukocytes in neoplastic tissue [7]. Currently, data in the literature confirm the existence of inflammation in the tumor microenvironment, which is consistent with the Virchow hypothesis, and the increased risk of cancer in cases of chronic inflammation. About 10% to 20% of cancers are related to chronic infections and the accompanying inflammation [8, 9]. Previous data indicate that the inflammatory microenvironment is an important element in all types of cancer, including those where a direct causal relationship with inflammation has not yet been confirmed [8, 10].
Thus, this work aimed to summarize the latest information on the relationship between the microbiome, inflammation, and carcinogenesis. For this purpose, a literature review was conducted, focusing on new achievements in research on the human microbiome, with particular emphasis on selected viruses, bacterial strains, and fungal species, and their impact on inflammation and carcinogenesis. More than 100 peer-reviewed scientific articles, conference materials, and short communications that had been published between 1881 and 2023 were analyzed, although the main focus was on articles from the last 10 years, primarily regarding viruses, bacteria, and fungi as proven carcinogens, the role of inflammation in carcinogenesis, and the possibilities of therapeutic use of pro- and prebiotics.
One of the groups of risk factors that may cause the development of cancer, apart from physical and chemical factors, are microbiological factors that usually precede the development of cancer, and acute and chronic inflammation [11, 12, 13]. Such factors include, among others, Helicobacter pylori predisposing to the development of gastric cancer, Human papillomavirus causing cervical cancer or hepatitis virus being conducive to the development of cancer of this organ [14, 15].
Hepatocellular carcinoma (HCC) is the most common type of liver cancer and the
third leading cause of cancer-related death worldwide. Among the chemical factors
predisposing to the development of HCC, alcohol is the most important, and the
risk of HCC increases at a level of consumption of 10 g of alcohol per day [16].
Another factor is smoking tobacco, which contains aromatic hydrocarbons,
diethylnitrosamine, and 4-aminobiphenyl [17]. Environmental factors contributing
to the occurrence of HCC include aflatoxins, vinyl chloride, arsenic compounds,
polychlorinated biphenyls, and radioactive compounds [18]. Previous data indicate
an extremely important role for the liver microenvironment in cancer development
and metastasis. The organ’s microenvironment plays a key role in the
proliferation and migration of cancer cells [19]. Various components of the
macroenvironment should be considered in the development of cancer and its
progression in the liver, whereby both proteins included in the extracellular
matrix (ECM), such as collagen, and other various cell types: bone-marrow-derived
macrophages, such as tumor-associated macrophages (TAMs), Kupffer cells (KCs),
liver sinusoidal endothelial cells (LSECs), immune cells like tumor-associated
neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), fibroblasts,
hepatocytes, and hepatic stellate cells (HSCs) [20, 21]. The ECM forms a framework
and contains domains enabling proteins with various functions to bind, although
it also contains proteins that interact with growth factors (including HGF:
hepatocyte growth factor; VEGF: vascular endothelial growth factor). These
factors promote cell migration and angiogenesis, which in turn enables metastatic
progression. In the initial stages of tumor development, the microenvironment
inhibits this process, yet at some point during tumor growth, the
microenvironment eventually supports this growth [22]. Although obesity and
alcohol abuse are the factors that predominantly contribute to the development of
this type of cancer, hepatitis B virus (HBV) or hepatitis C virus (HCV) infection
increases the chance of developing liver cancer by 15–17 times [23]. HCV and HBV
prevention programs, vaccinations, and modern forms of treatment are currently
changing the epidemiology of HCC, as the etiologies of non-viral diseases are
evidently increasing [24, 25]. Recent studies have shown that the translocation of
intestinal microorganisms that contribute to the development of inflammation in
the liver and its fibrosis, together with the activation of Toll-like receptors
(TLRs), stimulates the development of HCC [26]. Activation of TLRs, in particular
TLR-4, triggers the NF-
Many infectious agents in the liver microenvironment can destabilize the normal
functioning of liver cells, inter alia, by modulating components of the
Wnt/
In addition to avoiding recognition by the immune system, cancer cells induce
inflammation and create a favorable tumor microenvironment in order to progress
to full malignancy [35]. For this purpose, they use various mechanisms through
which oncogenic mutations provide cancer cells with the ability to dysregulate
mitogenesis, resistance to apoptosis, and the ability to invade and infiltrate
healthy tissues [36]. Signaling pathways that are disturbed during cancer
transformation include receptor tyrosine kinases (RTKs). In cancer, dysregulation
of RTK activation caused by oncogenic mutations is common [37]. Moreover, cancer
cells are insensitive to control mechanisms related to cadherin-mediated contact
inhibition, and dysregulated integrin signaling is important in the process of
tumorigenesis [38]. Other metabolic reprogramming pathways that are associated
with carcinogenesis include mutations in metabolic enzymes. Previous data
indicate the presence of heterozygous mutations in one of the two genes encoding
isocitrate dehydrogenase in glioma and leukemia cells [39]. Cell division is
regulated by the activation of cyclin-dependent kinases (CDKs), the catalytic
subunits of cyclins [40]. In various types of cancer, oncogenic mutations change
various elements in the signaling network of cyclins and CDKs, whereby, among
others, cyclin D1 is significantly overexpressed in breast cancer cells [41].
Research results published by Shi Y et al. [42] and Massague J et al. [43] indicate an
important role of transforming growth factor beta (TGF-
The T-cell response to viruses in patients with chronic infection and cirrhosis is delayed and transient compared to the strong response in those who are able to fight off the infection. Thus, although HCC can be induced by direct transformation of cells by HBV, tumor progression for both viruses is mainly dependent on the development of inflammation in the organ. Hepatitis caused by HBV has often been associated with intestinal dysbiosis, which is manifested by an increase in the number of fungal species and a simultaneous decrease in the number and diversity of strains belonging to the Bifidobacterium species. Studies on experimental animals have shown the influence of the intestinal microbiota on the progression of liver diseases and cancer in this organ [45, 46, 47]. When analyzing the impact of HBV and HCV on the possibility of HCC, the hepatitis delta virus (HDV) should also be considered. It is a small, defective virus that needs HBV to replicate and multiply effectively. Literature on the role of HDV infection in HBV-related liver disease or liver cancer is very limited; however, it suggests that HBV/HDV coinfection is associated with an increased risk of developing HCC [48].
The relationship between the direct transformation of cells by viruses and inflammation-induced carcinogenesis is also possible for many other oncogenic viruses. In addition to HBV and HCV, there are five other viruses with confirmed oncogenic effects on the human body: HPV, Merkel cell polyomavirus (MCPyV, MCV), human herpesvirus type 8 (HHV-8), which is the cause of sarcoma Kaposi and other conditions, especially in immunocompromised people due to various dysfunctions in their immune systems, the EBV, which causes Burkitt lymphoma and human T-cell lymphotropic virus type 1 (HTLV-1) (Fig. 1) [49].
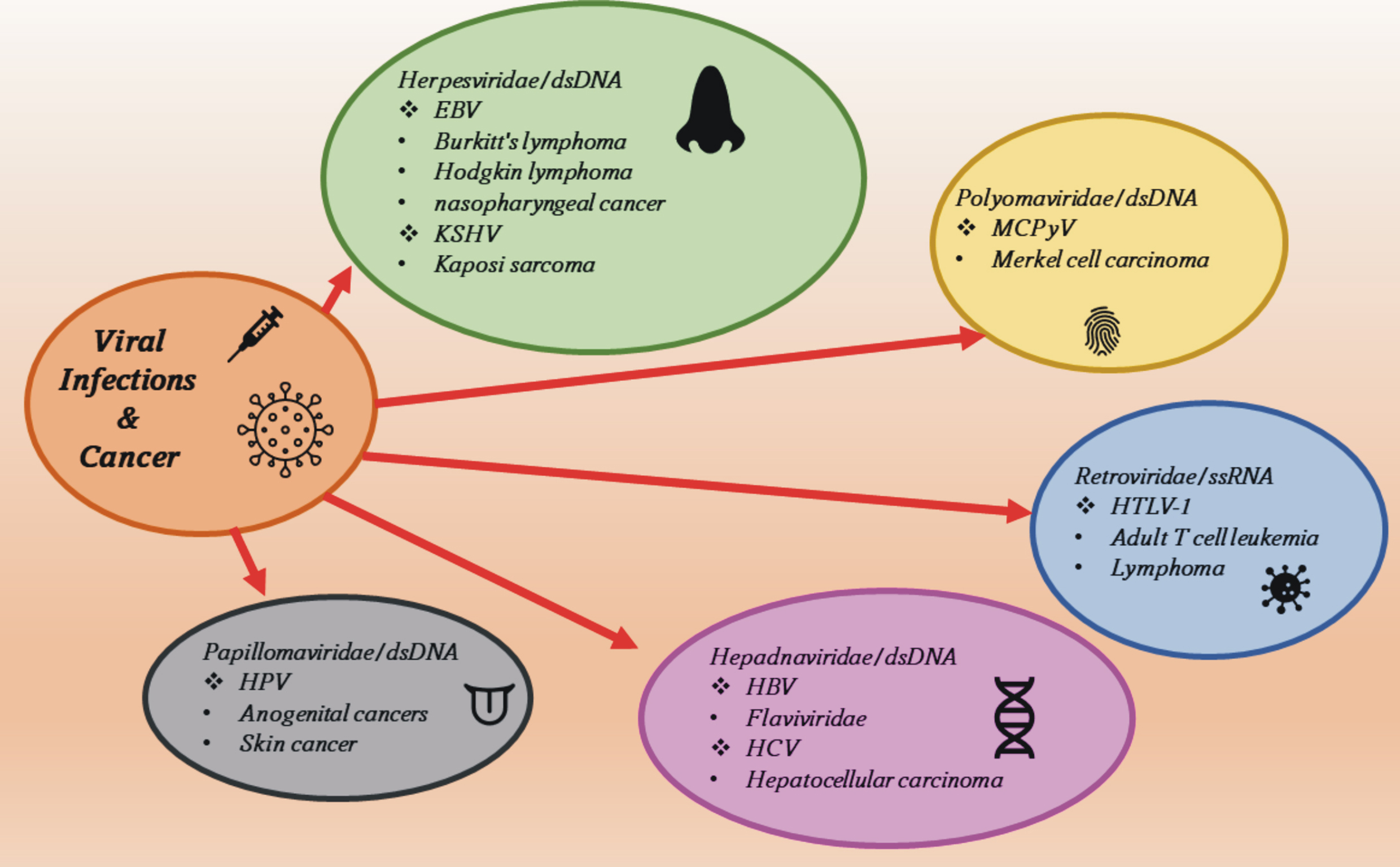 Fig. 1.
Fig. 1.Viral oncogenic agents and the diseases they cause. EBV, Epstein-Barr virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; HPV, Human papillomavirus; MCPyV, Merkel cell polyomavirus; HBV, hepatitis B virus; HCV, hepatitis C virus; HTLV-1, human T-lymphotropic virus type 1.
EBV is associated with the uncontrolled proliferation of B lymphocytes, T
lymphocytes, and natural killer (NK) cells. Malignant transformation can occur in
cells as a result of latent infection, and four types of EBV latency are known
that are associated with the expression of different proteins [50, 51]. Type I
latency is associated with Burkitt’s lymphoma, while type II latency is
associated with nasopharyngeal cancer, gastric cancer, Hodgkin’s lymphoma, and
T-cell and natural killer cell lymphoma. In turn, type III was found in people
with post-transplant lymphoproliferative disease and AIDS (Acquired Immune
Deficiency Syndrome) patients with diagnosed lymphoma [52, 53]. The KSHV (Kaposi’s
sarcoma-associated herpesvirus) virus infects B lymphocytes, macrophages,
monocytes, keratinocytes, and endothelial cells of blood vessels, and causes
Kaposi’s sarcoma alongside exudative lymphoma and multifocal Castleman’s disease
[54, 55]. The main oncoprotein in KSHV is the Lana protein (viral
latency-associated nuclear antigen), which causes the inhibition of cell
signaling pathways: the TGF-
The HPV virus infects epithelial cells causing skin, common, and flat warts,
however, some HPV strains can cause cervical, anal, vaginal, penile, and vulvar
cancers [63]. HPV viruses can be divided into five groups:
Although many bacterial strains play an unquestionable role in the processes of
carcinogenesis, Helicobacter pylori was classified by the International
Agency for Research on Cancer as a human class I carcinogen due to its proven
relationship with certain types of gastric cancer and lymphomas. H.
pylori can be considered a commensal microorganism and an opportunistic pathogen
because it is characterized by quite low virulence, while disease symptoms are
observed mainly in older people. This bacterium is identified in various body
fluids, and the human stomach is colonized early in life, usually in the family
environment. The factors that influence the development of diseases caused by H. pylori
are primarily diet, individual immunity, and the use of medications. All
these factors affect the balance of the microbiome, which also plays an important
role in H. pylori infections [73]. H. pylori has the ability to produce
urease, which helps it survive in the low pH of the stomach, and long-term
infection with this pathogen further raises the pH, making it easier for other
microorganisms to colonize the stomach. Long-term exposure to H. pylori
with an initial inflammatory response, including IL-1
Other oncogenic pathogenic microorganisms are some bacterial strains known to
promote the development of colorectal cancer, e.g., selected strains of
Escherichia coli, Streptococcus bovis, Bacteroides fragilis, Enterococcus
spp., and some species of the Enterobacteriaceae family (Fig. 2). They
can directly adhere to the intestinal epithelial layer and stimulate cell
proliferation, leading to hyperplasia. In addition, they synthesize enzymes and
toxins, such as 20 kDa heat-labile metalloproteinase, called B.
fragilis toxin (BFT), and adhesin FadA, a virulence factor identified
from Fusobacterium nucleatum, which violate the integrity of the
epithelial layer, damage cells, and cause inflammation [83, 84]. Of these species,
the most thoroughly studied in terms of its association with cancer is E.
coli. Its stimulating effect on the progression of colorectal cancers is
confirmed due to its adhesion to intestinal epithelial cells, which causes
hyper-proliferation and inflammation. Major virulence factors, mainly adhesins,
toxins, iron-acquisition factors, lipopolysaccharides, and invasins, are
carcinogenic through damage to DNA and epithelial/mucosal barriers [85].
Particularly pks+ strains that produce polyketide synthase are characterized by
high toxicity and an incidence associated with colorectal cancer. Enterotoxic
Bacteroides fragilis also generates toxins, as was mentioned above,
which lead to the development of colorectal cancer. They induce a variety of
responses at the molecular level, including cleavage of E-cadherins and
activation of
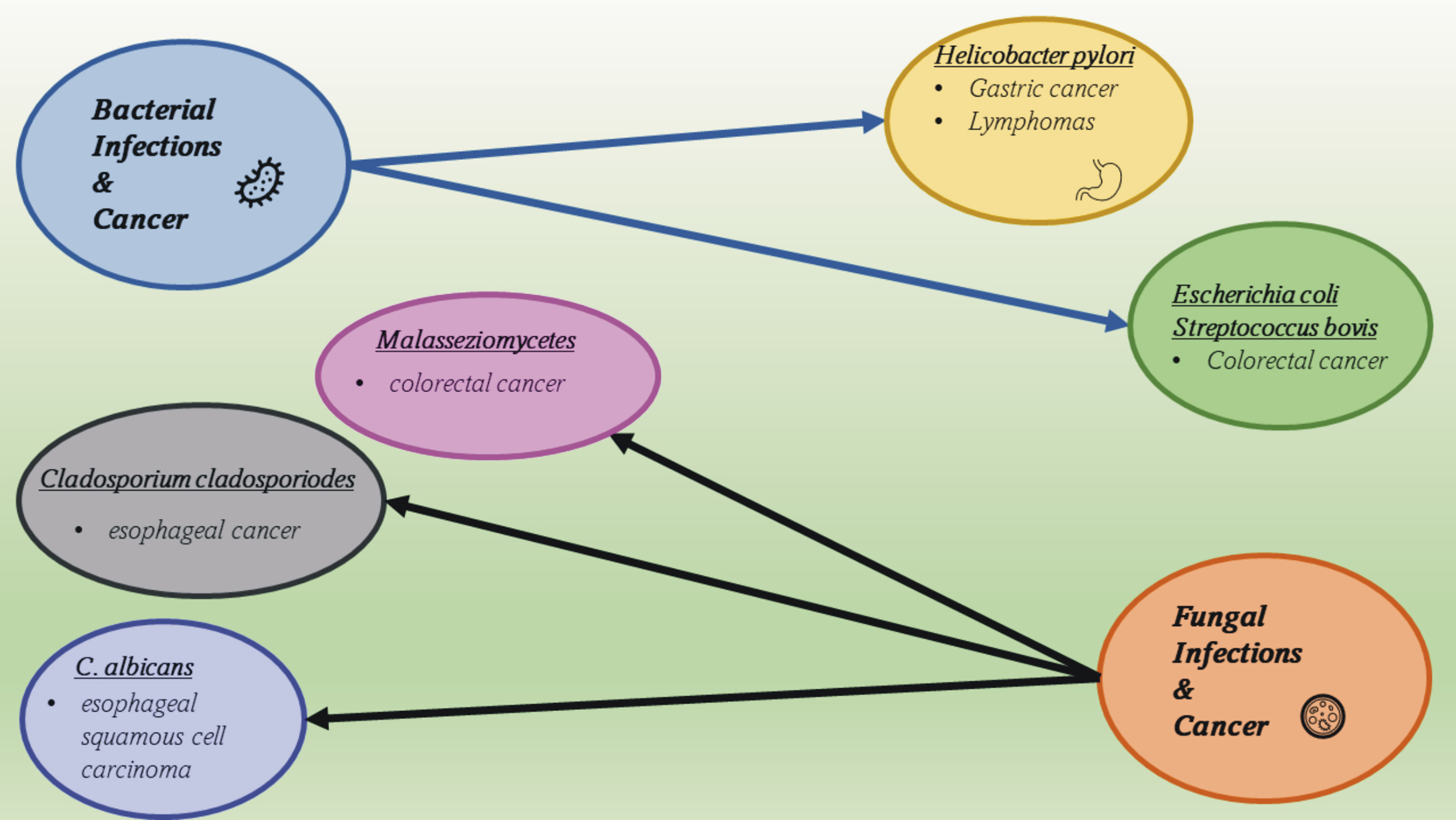 Fig. 2.
Fig. 2.Bacterial and fungal oncogenic agents and the diseases they cause.
Following the development of the research area related to analyzing the microbiome, fungi were found to be included in it and to also be important in the development of cancers [89]. Fungi that inhabit the human digestive system are characterized by high species variability and dependence on environmental factors, mainly diet [90]. They have been identified in the esophagus, stomach, pancreas, and intestines, while the most common genera are Candida, Saccharomyces, Malassezia, Cladosporum, Cryptococcus, Trichospora, and Aspergillus, among many others [91]. In cancers of the gastrointestinal tract, a disturbed ratio of Basidiomycota to Ascomycota has been reported. A particular overgrowth of Candida albicans was found in ESCC (esophageal squamous cell carcinoma), Cladosporium cladosporoides in esophageal tumors, and Malasseziomycetes in CRC (colorectal carcinoma) [92] (Fig. 2). In the case of fungi, as in the case of bacteria, pathogenic changes are usually caused by a disruption in the specific dynamic balance, leading to the occurrence of infections, often chronic ones. Fungi–bacteria interactions also seem to be key here, as they have been observed in cases of CRC. Disturbances in the composition and functioning of the microbiome are a way in which fungi influence the appearance and development of gastrointestinal cancers.
Chronic inflammation significantly predisposes the initiation of cancer but also favors its development, creating the inflammatory tumor microenvironment, where cells change their phenotypic and functional characteristics. In approximately 20% of cancer cases, the appearance of the tumor is preceded by infection and long-term inflammation [93]. Disturbances in microbiome homeostasis and interactions by microorganisms with hematopoietic cells are associated with both inflammation and carcinogenesis. In animal models, it has been shown that IL-18 has a protective function in relation to mucous membranes. Mice that are deficient in synthesizing and responding to IL-18 are characterized by intestinal dysbiosis and increased sensitivity to chemically induced colorectal cancer. Short-chain fatty acids (SCFAs), such as butyric acid, which is a product of bacterial fermentation of fiber in the intestine, induce the synthesis of IL-18 by intestinal epithelial cells by activating GPR109a receptors, yet also act directly on macrophages and T cells. SCFAs induce the expansion of Treg cells to produce anti-inflammatory cytokines, including IL-10, which inhibits the development of inflammation and carcinogenesis. IL-18 stimulates the repair of tissues, mainly mucosa, by regulating the production and increasing IL-22 availability through its production by intestinal lymphoid cells and, by activating STAT3, which induces epithelial cell proliferation and synthesis of antibacterial peptides. Therefore, IL-22, by stimulating the repair of damaged epithelia, may act, depending on the degree of damage, as a pro- or anti-carcinogen [94, 95].
In inflammation caused by pathogenic microorganisms that contribute toward the
development of cancer, the accumulation of reactive oxygen and nitrogen species
plays an important role. Both types of free radicals cause the formation of
cross-links in DNA and breakage of DNA and RNA strands, which promotes genome
instability and mutations in oncogenes and tumor suppressor genes. Inflammatory
conditions in the body create conditions conducive to the development of the
tumor microenvironment, in which the decisive role is played by hematopoietic
cells, such as macrophages, which, under the influence of tumor cells secreting,
among others, IL-10 and TGF-
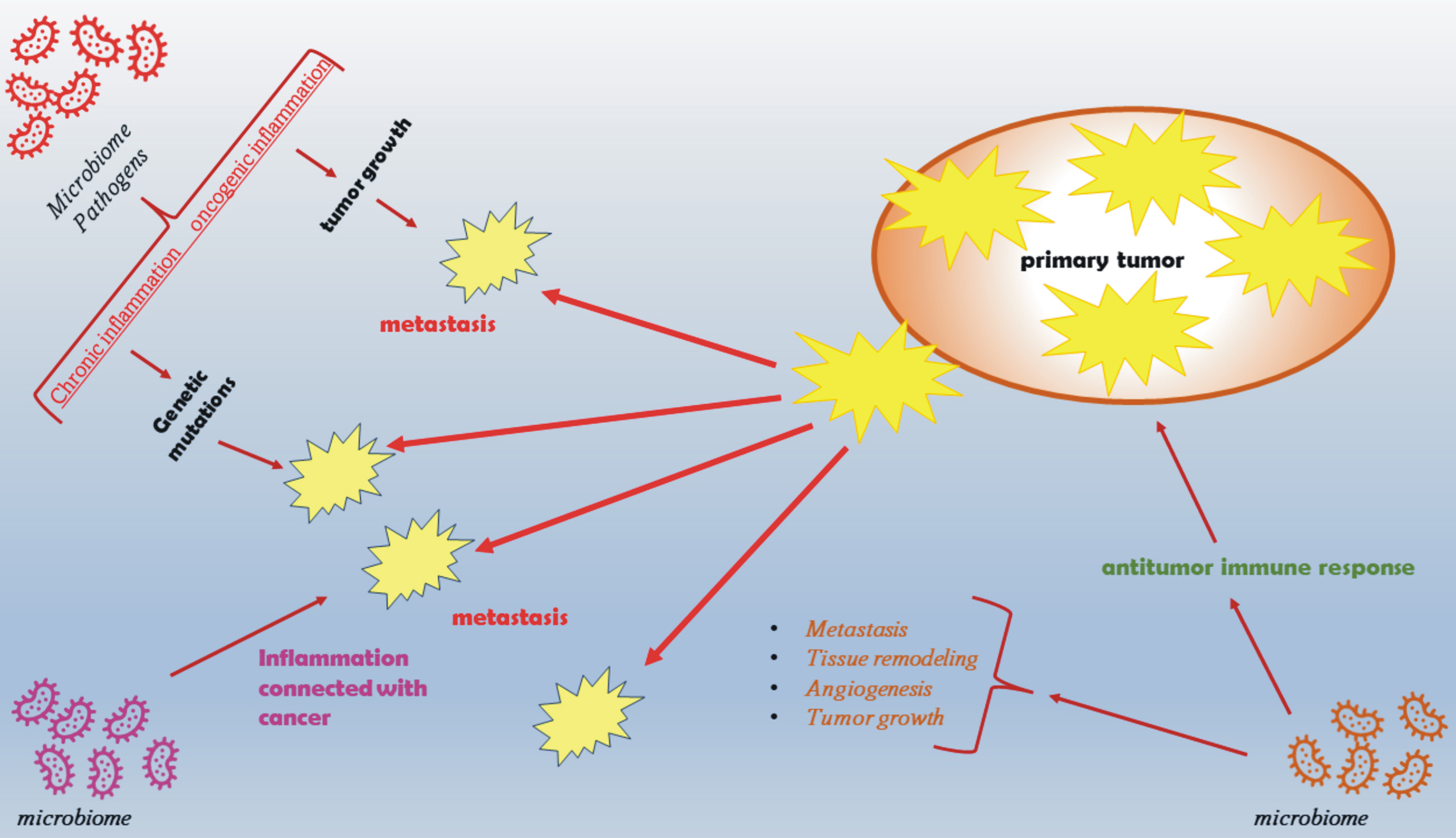 Fig. 3.
Fig. 3.The role of inflammation and the microbiome in tumor initiation, promotion, metastasis, and response to therapy. The microbiome directly and indirectly influences the development and maintenance of inflammation in the body, which in turn stimulates the development of cancers by increasing genetic instability and by creating a microenvironment conducive to tumor growth. The microbiome may also influence the emergence of cancer-promoting factors, such as obesity and metabolic syndrome, and modulate the immune response mechanisms that regulate tumor initiation and progression.
Tumor-associated macrophages (TAM macrophages) produce pro-angiogenic factors
(VEGF-A, TNF-
A better understanding of the mechanisms involved in cancer development at the molecular level will allow the inclusion of therapeutic factors, such as pro- and prebiotics, the better use of antibiotics and manipulations at the level of individual bacterial proteins to enable or disable specific factors responsible for the toxicity and carcinogenicity of selected bacterial strains (Fig. 4) [100]. When considering the impact of the microbiome on carcinogenesis, we should also mention the exposome theory, the impact on the human body, which is crucial. The exposome is composed of many factors interacting (Fig. 4). The exposome can be defined as an integrated function of the exposure of the human body, which includes everything that surrounds us, the place we live in, the relationships we create, the food we consume, the drugs we use, and the activities we perform. It is worth noting that exposure is not limited to the substances or stimulants that we consume but also includes chemical compounds produced in the body in metabolic processes [101]. Therefore, the microbiome and its functioning and impact are important components of the exposome, which to some extent can reduce the unfavorable impact of selected elements of the exposome on the human body.
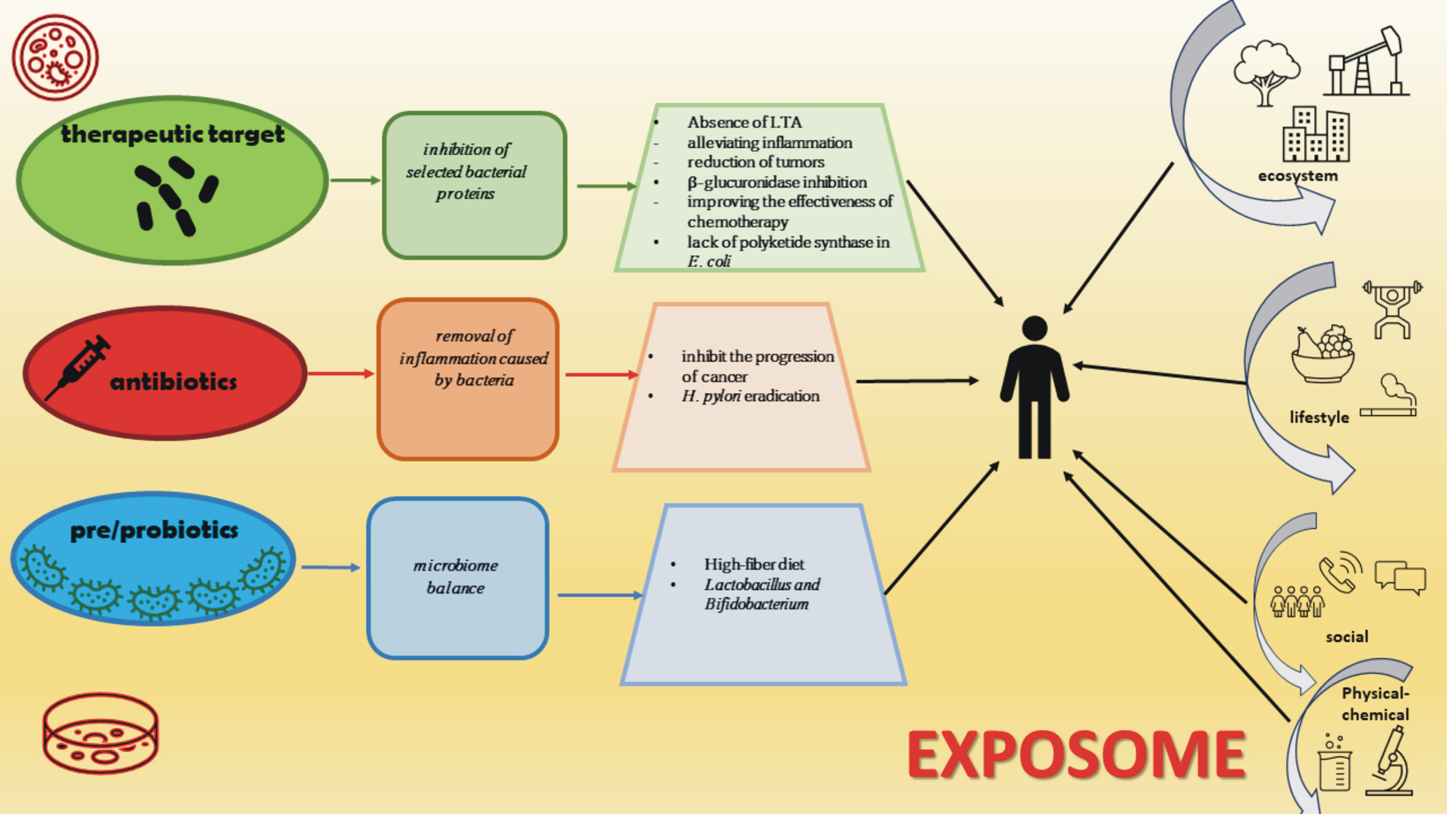 Fig. 4.
Fig. 4.The microbiome, cancer, and exposome. Three potential interventions point to improved patient responses to cancer therapy and the impact of the exposome on the human body.
In recent years, the following have become very popular:
Both are used in the prevention and treatment of certain diseases, mainly in the gastrointestinal tract and have even become a key element in the daily diet. There have been reports showing the positive effect of dietary fiber on the synthesis of SCFAs by intestinal bacterial strains and its possible beneficial effect on cancer cells. In addition, the consumption of large amounts of fiber increases methanogenesis, which leads to a reduction in the number of hydrogen-producing bacteria, which is extremely important because excess hydrogen in the intestine prevents the regeneration of NAD (nicotinamide adenine dinucleotide). Another beneficial aspect resulting from the consumption of large amounts of fiber is the colonization of the intestines by microorganisms that eliminate possible inflammation, e.g., Faecalibacterium prausnitzii. The most commonly used probiotic strains are Lactobacillus and Bifidobacterium spp., which enhance the detoxification of toxic metabolites and carcinogens in the intestines, thereby stimulating the body’s anticancer response and producing anticancer and antimutagenic components that directly interact with cancer cells, inhibiting their growth, and synthesizing substances such as SFCAs that are important for maintaining the immune balance [102, 103].
Although probiotics are very intensively studied, therapeutic strategies also
include pathogenic microorganisms. There are animal studies that have shown the
ability to inhibit specific bacterial proteins without disturbing the microbial
balance of the host. For example, animals infected with a strain of
Lactobacillus acidophilus and lacking lipoteichoic acid (LTA-) showed
milder inflammation of the colon and inhibition of intestinal tumor growth.
Another group of researchers showed the alleviation of intestinal inflammation
associated with cancer after infection with the E. coli pks- strain in
an animal model. Cyclomodulins, e.g., colibactin (pks), are virulence
factors that modulate cellular differentiation, apoptosis, and proliferation. The
depletion of bacteria with selected proteins, such as pks, can alleviate the
symptoms of the disease. The effects of manipulating bacterial enzymes on human
health were also studied. The anticancer drug irinotecan causes constant diarrhea
in patients, which limits the possibilities of effective therapy. Wallace
et al. [104] developed inhibitors for the bacterial enzyme
Overall, it should be said that the impact of microorganisms on the functioning of the human body is huge and indisputable. The microbiome can indeed be considered a separate “organ”. Both pathogenic and commensal bacteria actively participate in the formation of inflammation and the development of cancer in humans. Moreover, external and environmental factors, such as diet, consumed drugs, and toxins have an impact on the composition and function of the human microbiome, which can lead to dysbiosis and tissue homeostasis disorders through various mechanisms, including the enhancement of the immune response, activation of epithelial proliferation, and impairment of barrier integrity. Currently, although science is at the stage of revealing the complexity of the mechanisms involved in building the microbiome-inflammation-cancer relationship, we already know that taking care of the body’s microbial homeostasis is as important as any other organ in our body.
AJT designed the research study, analyzed the information, and wrote and edited the manuscript. AJT contributed to editorial changes in the manuscript. AJT read and approved the final manuscript. AJT has participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was financially supported by the Ministry of Education and Science, Poland, under the research project number WZ/WB-IIŚ/6/2022.
The author declares no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.