
An autoimmune reaction directed against the cardiac b1-adrenergic receptor (ß1-ADR) leading to the generation of autoantibodies (AA) against this G-coupled receptor has been described in patients with heart failure (HF). Agonist-like ß1-ADR-AA are associated with morbidity in HF patients and even predict mortality. Standardised and valid diagnostic tools to detect ß1-ADR-AA in clinical routine are lacking. We used a novel ELISA approach to investigate ß1-ADR-AA in a cohort of 574 HF patients of the CIBIS-ELD trial with follow up. The CIBIS-ELD trial compared the titration of bisoprolol and carvedilol to recommended target doses in regard to BB tolerability in patients aged 65 years and older. Patient with left ventricular (LV) ejection fraction (EF) < 50% or LV diameter end diastolic (DED) >55 cm showed significantly higher levels of ß1-ADR-AA. Although not yet fully validated, this ELISA allowed for a negative correlation of ß1-ADR-AA with the EF at baseline and at the follow up, ß1-ADR-AA further correlated positively with basal heart rate at follow up 12 weeks later. ß1-ADR-AA levels thus determined significantly increased under titration with beta-blockers (p<0.0.1). Changes in ß1-ADR-AA between F-Up and baseline were significantly higher in patients who used beta blockers (p=0.0.16) before study inclusion. The type of beta-blocker (BB) titrated in this study did not affect log ß1-ADR-AA levels at baseline (p=0.1.32), follow-up (p=0.0.58), nor the change (p=0.4.26). ß1-ADR-AA levels were estimated using a novel, commercially available ELISA. Although not yet fully validated, this ELISA allowed for pathophysiological insights: ß1-ADR-AA levels thus determined significantly increased under titration with beta-blockers (p<0.0.1), irrespective of type of BB. Higher levels of ß1-ADR-AA at baseline are associated with higher heart rates, lower ejection fraction and enlarged left ventricles. The relevance of the ß1-ADR-AA biomarker should be further evaluated.
ß1-Adrenoreceptors (ß1-ADR) mediate the positive inotropic, chronotropic, bathmotropic, dromotropic, and lusitropic effects of endogenous neuro-hormonal stress on cardiac tissues (1), (2). Beta Blockers (BB) shield the heart from such catecholaminergic stress – i.e. by blockade of the cardiac ß1-ADR - and have emerged as a cornerstone a of optimum adjusted medical therapy of Heart Failure (HF) (3) and remain of paramount importance in the routine clinical treatment of patients suffering from this debilitating syndrome.
Recent advances in the understanding of HF pathophysiology suggest a contributing role of autoimmune mechanisms in the development and progression of HF (4). Autoimmune reactions directed against the cardiac ß1-ADR may lead to the generation of autoantibodies (ß1-ADR-AA) against this G-coupled receptor (5). Molecular mimicry i.e. leads to ß1-ADR-AA in patients with Chagas cardiomyopathy (6), (7). ß1-ADR-AA may interfere with ligand binding and receptor conformation, i.e. increasing cAMP stimulation (8) or amplifying the effect of catecholamines (9).
ß1-ADR-AA have previously been associated with cardiac dilatation as well as pump failure (10) in dilatative (DCM) (10), (11), (8) and ischemic cardiomyopathy (ICM) (10) but not necessarily in HF of other aetiologies (11). Aside from association, agonistic-like ß1-ADR-AA were found to predict mortality in idiopathic cardiomyopathy (12). Passive transfer of agonist-like ß1-ADR-AA induced heart failure in rodents (13). These findings indicate the specific role of such AA in the pathophysiology and development of HF and have furthermore opened a window of opportunity of therapeutic approaches in our patients. Removing the AA from the bloodstream by means of immunoadsorption - as a non-pharmacologic concept - has already shown to significantly improve cardiac performance and clinical status in patients (14), (15). Pharmacologic concepts such as aiming at the partial abrogation of ß1-ADR-AA by use of cardio selective BB (16), and binding and neutralizing of ß1-ADR-AA by aptamers (17), or the cyclic peptide COR-1 (18) have been reported.
Furthermore, diagnostic and therapeutic implications have recently received growing attention in the HF community (4). Standardised and valid assessments of ß1-ADR-AA may prove pivotal for an understanding of pathophysiology, diagnosis, and risk-stratification in HF patients. In a situation of interference of ß1-ADR-AA with established, life-saving BB therapies, an option to monitor therapy response may be required. Autoimmune diseases such as grave’s or myasthenia gravis have taught us that the development of suitable assays is especially complex in the setting of autoimmunity (19). ß1-ADR-AA were measured by intracellular cAMP by fluorescence resonance energy transfer using a highly sensitive cAMP sensor, a very sensitive method which is not suitable in clinical routine (20). Recently a cell-based competition ELISA has been introduced (21). Hence, to prospectively address cardiac autoimmunity new diagnostic studies (i.e. ETiCS (22)) are currently recruiting.
In the present analysis, we used a solid-phase ELISA for the detection of antibodies (IgG) targeted against the g-protein-coupled ß1-adrenoreceptor from baseline and follow-up blood samples collected within the CIBIS-ELD (23) trial on selective vs. non-selective beta-blocker titration in HF patients. The ELISA used within this analysis is (i) not a peptide-ELISA, (ii) it is not yet fully validated regarding sensitivity and specificity compared to functional bioassays, and (iii) should therefore be considered a putative surrogate marker for b1ARAAB. In this secondary post-hoc analysis we investigated the association between ß1-ADR-AA - as the novel Biomarker – and HF characteristics, severity and possible implications of beta-blocker therapy. The present study serves to provide clinical correlation for this marker.
The CIBIS-ELD trial design (24) and its primary results (23) have been reported previously. In principle, the trial compared the titration of bisoprolol and carvedilol to recommended target doses in regard to BB tolerability. Patients aged 65 years and older with clinical, chronic stable heart failure with either reduced or preserved ejection fraction were randomized to either BB. At baseline all patients were BB naïve or receiving 25% or less of the target dose. BB dose was titrated according to a standardized protocol every two weeks over a 12-week period, starting with 1.2.5 mg bisoprolol daily or 3.1.25 mg carvedilol twice daily. Target doses were 10 mg bisoprolol daily or 25 mg carvedilol twice daily. Adverse events were recorded at every visit; treatment-related events were defined according to a blinded committee.
Data on demographics, medical history, electrocardiogram, echocardiography, New York Heart Association (NYHA) functional class, and laboratory parameters were collected at baseline and after up-titration at 12 weeks. Glomerular filtration rate (GFR) was calculated using the MDRD formula. Serum ß1-ADR-AAs were measured with a commercially available sandwich ELISA (CellTrend GmbH Luckenwalde, Germany) according to the instruction manual. The microtiter 96-well polystyrene plates were coated with extracts from transfected Chinese Hamster Ovary (CHO) cells overexpressing the human ß1-ADR. To conserve conformational epitopes of the receptor as much as possible, a concentration of 1 mM calcium chloride was maintained during all steps of the assay. Duplicate samples of a 1:100 serum dilution were incubated at 48°C for 2 hours. After washing steps, plates were incubated for 60 minutes with a 1:20 000 dilution of horseradishperoxidase-labeled goat anti-human IgG (Jackson, Bar Harbor, ME, USA) used for detection. In order to obtain a standard curve, plates were incubated with test sera from an anti-ß1-ADR antibody positive index patient. All tests were done in duplicate. The detection threshold of ß1-ADR-AA was set at 2.5. U. All sera were coded and assessed for ß1-ADR-AA by individuals who had no information regarding the patients’ characteristics.
This is secondary a post-hoc analysis. Baseline variables are presented as frequencies and percentages for binary variables, or means and standard error (SE), i.e. in combination with a median-split by log-transformed ß1-ADR-AA. Data are presented overall and separately in the respective groups. Comparisons of all groups were performed by Fisher’s exact test or Wilcoxon test, respectively. For beta blockers, heart rate, ECG, systolic and diastolic blood pressure, 6 min walk test, NYHA class and echocardiographic parameters, the relationships between pre- and post-titration measurement were examined. Quantitative measures were analyzed by Kandall’s rank correlation. R (R Core Team (2015). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/.) was used as statistical software.
The CIBIS-ELD trial (ISRCTN 34827306 at controlled-trials.com) was approved by the institutional ethical review boards of all participating centres. The study conforms with the principles outlined in the Declaration of Helsinki. All patients provided written informed consent prior to enrolment.
CIBIS-ELD included 876 patients in the primary analysis (431 bisoprolol, 445 carvedilol) and 782 in the follow-up analysis (386/396) (23). We report here on a sample of 574 patients (286 bisoprolol, 288 carvedilol) of the CIBIS-ELD trial population with available serum analysis. Baseline characteristics of the population according to a median split of log ß1-ADR-AA levels are displayed in Table 1.
| Variable | VarN° of patients | all patients descriptive Value | N° of patientslow ß1-AAK | low ß1-AAKdescriptive Value | N° of patientshigh ß1-AAK | high ß1-AAKdescriptive Value | high vs. lowp= |
|---|---|---|---|---|---|---|---|
| (n=) | (mean±sd; percent) | (n=) | (mean±sd; percent) | (n=) | (mean±sd; percent) | ||
| Demographics & Heart Failure | |||||||
| Age [years] | 574 | 72±5.4 | 287 | 73±5.5 | 287 | 72±5.4 | 0.083 |
| Sex [female] | 574 | 198 (34%) | 287 | 111 (39%) | 287 | 87 (30%) | * 0.043 |
| Systolic blood pressure [mmHg] | 574 | 138±21.3 | 287 | 140±21.3 | 287 | 136±21.1 | * 0.037 |
| Diastolic blood pressure [mmHg] | 574 | 81±11.7 | 287 | 81±11.6 | 287 | 81±11.7 | 0.671 |
| LVEF < 50 % | 569 | 379 (67%) | 286 | 175 (61%) | 283 | 204 (72%) | ** 0.006 |
| NYHA Class | |||||||
| I | 574 | 21 (4%) | 287 | 11 (4%) | 287 | 10 (3%) | 0.607 |
| II | 392 (68%) | 201 (70%) | 191 (67%) | ||||
| III | 161 (28%) | 75 (26%) | 86 (30%) | ||||
| NT-pro-BNP quantile | 545 | 270 | 275 | ||||
| 1st | 137 (25%) | 81 (30%) | 56 (20%) | 0.060 | |||
| 2nd | 136 (25%) | 59 (22%) | 77 (28%) | ||||
| 3rd | 136 (25%) | 65 (24%) | 71 (26%) | ||||
| 4th | 136 (25%) | 65 (24%) | 71 (26%) | ||||
| Heart Failure Etiology | |||||||
| Hypertension | 563 | 194 (34%) | 282 | 106 (38%) | 281 | 88 (31%) | 0.333 |
| Coronary Artery Disease | 261 (46%) | 126 (45%) | 135 (48%) | ||||
| Cardiomyopathy | 44 (8%) | 18 (6%) | 26 (9%) | ||||
| Other | 64 (11%) | 32 (11%) | 32 (11%) | ||||
| Peripheral Edema | 573 | 107 (19%) | 287 | 66 (23%) | 286 | 41 (14%) | * 0.014 |
| 6 Minute Walk Test walk distance [m] | 538 | 332±106 | 266 | 332±103 | 272 | 331±109 | 0.913 |
| Hospitalization for heart failure during the past 12 months | 572 | 202 (35%) | 285 | 90 (32%) | 287 | 112 (39%) | 0.067 |
| Quality of life | 494 | 246 | 248 | ||||
| SF-36 physical score | 39±9.7 | 39±9.8 | 38±9.5 | 0.628 | |||
| SF-36 psychosocial score | 46±11.9 | 46±11.7 | 46±12.2 | 0.918 | |||
| Medical History | |||||||
| Atrial Fibrillation | 563 | 261 (46%) | 282 | 126 (45%) | 281 | 135 (48%) | 0.333 |
| Arterial Hypertension | 574 | 481 (84%) | 287 | 253 (88%) | 287 | 228 (79%) | ** 0.006 |
| Hyperlipoproteinemia | 573 | 370 (65%) | 287 | 196 (68%) | 286 | 174 (61%) | 0.067 |
| Diabetes mellitus | 574 | 153 (27%) | 287 | 84 (29%) | 287 | 69 (24%) | 0.186 |
| Renal dysfunction [GFR < 60ml] | 553 | 211 (38%) | 278 | 105 (38%) | 275 | 106 (39%) | 0.861 |
| Hyperuricemia | 574 | 51 (9%) | 287 | 27 (9%) | 287 | 24 (8%) | 0.770 |
| COPD | 574 | 45 (8%) | 287 | 26 (9%) | 287 | 19 (7%) | 0.352 |
| pAVD | 573 | 37 (6%) | 286 | 24 (8%) | 287 | 13 (5%) | 0.064 |
| Smoking | 574 | 287 | 287 | ||||
| Non-smoker | 326 (57%) | 163 (57%) | 163 (57%) | 0.215 | |||
| Ex-smoker | 199 (35%) | 105 (37%) | 94 (33%) | ||||
| Smoker | 49 (9%) | 19 (7%) | 30 (10%) | ||||
| Anthropometrics | |||||||
| Weight [kg] | 574 | 78±14 | 287 | 78±13.4 | 287 | 79±14.6 | 0.414 |
| Waist [cm] | 552 | 100±13.2 | 269 | 100±12.6 | 283 | 100±13.8 | 0.808 |
| Hip [cm] | 552 | 106±10.9 | 269 | 106±10.9 | 283 | 106±10.9 | 0.974 |
| BMI [kg/m2] | 574 | 27.8±4.6 | 287 | 28±4.6 | 287 | 27.7±4.6 | 0.430 |
| Transthoracic Echocardiography | |||||||
| Left ventricular ejection fraction [%] | 574 | 42±13.7 | 287 | 43±13.9 | 287 | 40±13.3 | * 0.010 |
| LVDED [mm] | 569 | 56±10.2 | 285 | 55±9.9 | 284 | 57±10.5 | ** 0.009 |
| LVDES [mm] | 556 | 43±11.6 | 276 | 41±11.1 | 280 | 44±11.8 | ** 0.001 |
| IVSED [mm] | 569 | 11.3±2.61 | 285 | 11.5±2.79 | 284 | 11±2.39 | * 0.017 |
| PWED [mm] | 568 | 10.6±2.29 | 285 | 10.8±2.42 | 283 | 10.3±2.13 | * 0.013 |
| E-Wave [cm/s] | 535 | 77±45 | 272 | 77±48 | 263 | 77±43 | 0.917 |
| e´-Wave [cm/s] | 504 | 7.8±4 | 259 | 7.7±4.2 | 245 | 8±3.9 | 0.357 |
| E/e’ | 502 | 11.9±8.7 | 259 | 11.9±8.2 | 243 | 11.9±9.2 | 0.977 |
| LAVI [ml/m2] | 528 | 34±15.4 | 267 | 34±15.6 | 261 | 33±15.3 | 0.561 |
| Deceleration Time [ms] | 529 | 220±86 | 269 | 219±86 | 260 | 221±85 | 0.830 |
| Fractional shortening [%] | 556 | 25.6±10.6 | 276 | 26.7±10.7 | 280 | 24.4±10.3 | * 0.010 |
| Elektrocardiogram | |||||||
| Heart Rate [bpm] | 574 | 73±14.6 | 144 | 71±14.2 | 144 | 74±15 | * 0.047 |
| Sinusrhythm | 563 | 194 (34%) | 282 | 106 (38%) | 281 | 88 (31%) | 0.333 |
| PQ interval [ms] | 449 | 171±33 | 233 | 172±33 | 216 | 169±33 | 0.265 |
| QRS interval [ms] | 572 | 104±26.9 | 286 | 104±26.2 | 286 | 104±27.7 | 0.977 |
| QT interval [ms] | 570 | 393±53 | 285 | 397±47 | 285 | 389±58 | + 0.050 |
| Left Bundle Branch Block | 563 | 74 (13%) | 281 | 37 (13%) | 282 | 37 (13%) | 1.000 |
| Laboratory Markers | |||||||
| Serum sodium [mmol/l] | 539 | 141±3.6 | 272 | 141±3.7 | 267 | 141±3.6 | 0.633 |
| Serum potassium [mmol/l] | 541 | 4.4±0.51 | 274 | 4.4±0.49 | 267 | 4.4±0.53 | 0.870 |
| Creatinine [mg/dL] | 553 | 101±34 | 278 | 99±29.2 | 275 | 103±38 | 0.162 |
| HbA1c [%] | 286 | 6.5±1.15 | 146 | 6.5±1.05 | 140 | 6.4±1.24 | 0.517 |
| N-terminal Propeptide BNP [pg/mL] | 545 | 1404±2543 | 270 | 1301±2131 | 275 | 1506±2892 | 0.348 |
| hs-CRP [mg/dL] | 428 | 0.52±0.79 | 207 | 0.42±0.58 | 221 | 0.61±0.93 | * 0.017 |
| log ß1-ADR-AA [U/mL] | 574 | 0.99±0.283 | 287 | 0.77±0.142 | 287 | 1.21±0.208 | ** 0.001 |
| Beta blocker therapy | |||||||
| Pre-treated with beta blocker [yes] | 574 | 348 (61%) | 287 | 170 (59%) | 287 | 178 (62%) | 0.550 |
| + P= 0.05, * P<0.05, ** P<0.01 | |||||||
At baseline, the proportion of women in the group with lower ß1-ADR-AA values was significantly higher than in the setting of higher ß1-ARD-AA, while age distribution was found to be similar in both groups. Regarding the clinically deemed aetiology of HF, NYHA functional class, 6-minutes-walk-test distance, NT-pro-BNP levels and history of HF hospitalization in the past 12 months, we report a similar clinical profile in the respective groups with lower or higher ß1-ADR-AA levels.
Patients with higher levels of baseline log ß1-ADR-AA had a significantly lower left ventricular (LV) ejection fraction (EF). When LVEF was classified categorically according to reduced vs. normal (LVEF ≤/> 55, p=0.0.035) and severely impaired vs. preserved (LVEF ≤35/≥50%, p=0.0.05) significant differences in log ß1-ADR-AA came to display (Figure 1). Similar results were observed in regard to LV end diastolic (DED) >55 cm (p=0.0.012) (Figure 2), and systolic (SED) diameter, where dilatation was seen in the group with higher levels of ß1-ADR-AA. Comparable results were observed for end-diastolic intra-ventricular septum thickness (IVSED) < 11mm (p=0.0.113) and end-diastolic posterior wall thickness < 11 mm (p=0.0.140) at baseline, follow-up and change. Thinner left ventricular walls were described when higher levels of log ß1-ADR-AAK were present.
 Figure 1.
Figure 1.
log ß1-ADR-AA classified according to LVEF ≤/> 55% (p=0.0035)
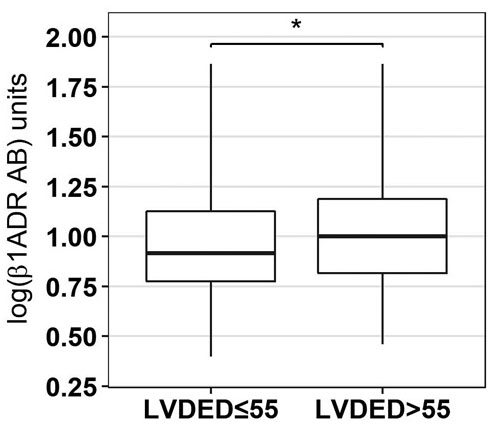 Figure 2.
Figure 2.
log ß1-ADR-AA classified according to LVDED) ≤/> 55 mm (p=0.0012)
Heart rate was lower in patients with lower ß1-ADR-AA levels. We report a significant association between baseline ß1-ADR-AA and resting heart rate (from ECG) at baseline (Table 2), at follow up 12 weeks later (r=0.1.41, p= 0.0.04) as well as in regard to change in HR during the study period (r=0.0.99, p=0.0.45). Change in ß1-ADR-AA did not correlate with change in HR. Systolic blood pressure tended to be higher in a situation of lower ß1-ADR-AA levels at baseline.
| Variable | Baseline [τ] | Baseline [p] | Change [τ] | Change [p] |
| Weight [kg] | 0.015 | 0.589 | -0.017 | 0.538 |
| Systolic blood pressure [mmHg] | - 0.090 | ** 0.003 | 0.047 | 0.336 |
| Diastolic blood pressure [mmHg] | - 0.037 | 0.208 | 0.019 | 0.541 |
| 6 Minute Walk Test walk distance [m] | - 0.006 | 0.823 | 0.044 | 0.381 |
| Transthoracic Echocardiography | ||||
| Left ventricular ejection fraction [%] | - 0.116 | ** 0.001 | 0.034 | 0.526 |
| LVDED [mm] | 0.072 | * 0.011 | -0.086 | 0.107 |
| LVDES [mm] | 0.088 | ** 0.001 | -0.093 | 0.093 |
| IVSED [mm] | - 0.044 | 0.135 | 0.031. | 0.598 |
| PWED [mm] | - 0.057 | 0.054 | -0.05 | 0.369 |
| LAES [mm] | - 0.050 | 0.082 | 0.112 | * 0.036 |
| Tissue Doppler E-Wave | 0.047 | 0.119 | -0.123 | * 0.022 |
| Tissue Doppler A-Wave | - 0.087 | ** 0.007 | -0.14 | * 0.015 |
| Deceleration Time [ms] | 0.011 | 0.695 | -0.005 | 0.921 |
| Electrocardiogram | ||||
| ECG Heart Rate [bpm] | 0.062 | * 0.028 | -0.011 | 0.818 |
| PQ interval [ms] | - 0.100 | 0.757 | -0.013 | 0.835 |
| QRS interval [ms] | 0.011 | 0.696 | -0.119 | * 0.024 |
| QT interval [ms] | - 0.049 | 0.082 | -0.05 | 0.339 |
| Laboratory Markers | ||||
| HbA1c [%] | - 0.011 | 0.788 | -0.029 | |
| N-terminal Propeptide BNP [pg/mL] | 0.052 | 0.072 | -0.098 | 0.097 |
| hs-CRP [mg/dL] | 0.060 | 0.065 | 0.033 | 0.697 |
| *P<0.05, ** P<0.01 | ||||
In a clinical models of heart failure with reduced (HFrEF, LVEF < 50%) and preserved ejection fraction (HFpEF, LVEF >50% and NTproBNP >125 (pg/mL)), we observed higher ß1-ADR-AA levels in HFrEF (1.0.5±0.2.77 vs. 0.9.5±0.3.3, p=0.0.13).
ß1-ADR-AA levels partially differed between different etiologies of heart failure. Hypertension and coronary artery disease (0.9.6±0.2.8 and 1.0.2±0.2.86, p=0.0.285) showed significantly different ß1-ADR-AA levels. Hypertension and cardiomyopathy (0.9.6±0.2.8 and 1.0.2±0.2.53, p=0.0.979) or coronary artery disease and cardiomyopathy (1.0.2±0.2.86 and 1.0.2±0.2.53, p=0.6.789) did not show statistically significant differences.
We observed a significant increase of ß1-ADR-AA levels under titration with beta-blockers (p<0.0.1) between baseline and after 12 weeks of follow-up (Figure 3). Levels of ß1-ADR-AA were independent of pre-use of beta blockers at the time of inclusion into the study. Changes in ß1-ADR-AA between follow-up and baseline were significantly higher in patients who used beta blockers (p=0.0.16) before study inclusion. The type of beta-blocker titrated in this study did not affect log ß1-ADR-AA levels at baseline (p=0.1.32), follow-up (p=0.0.58), nor the change (p=0.4.26). Further, in repeated measurement ANOVA, the type of beta-blocker titrated did not affect log ß1-ADR-AA levels (p=0.0.81), nor their change (p=0.4.09), while sex influenced values (p=0.0.05), but not change (p=0.6.28). Beta–blocker dosage did not affect ß1-ADR-AA levels.
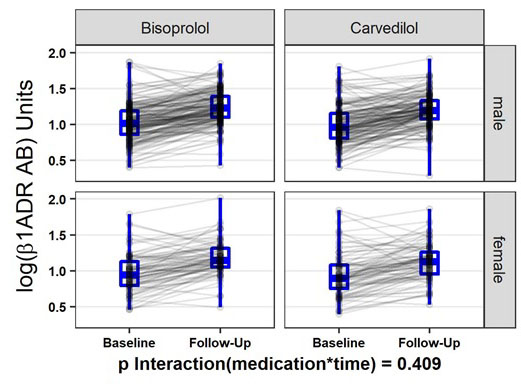 Figure 3.
Figure 3.
Repeated measurement ANOVA for change in log-ß1-ADR-AA by sex and Medication
Graphical display of correlation between log ß1ADR AB and HR, LVEF, LVDED and systolic blood pressure are provided in Figure 4-7.
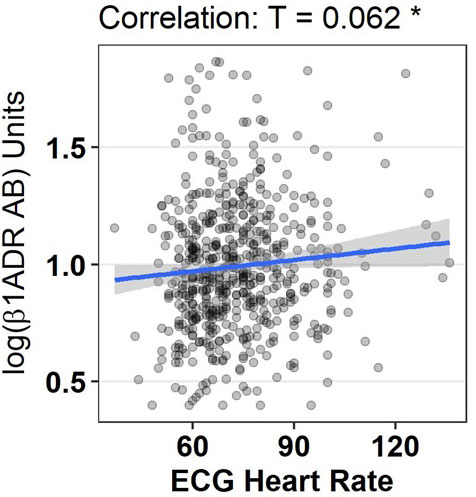 Figure 4.
Figure 4.
Graphical display of correlation between log ß1ADR AB and heart rate obtained by electrocardiogram
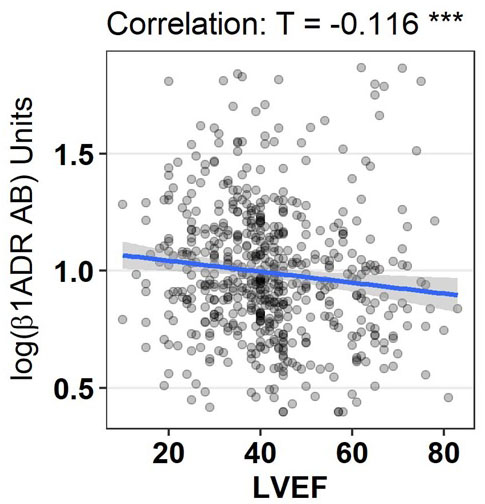 Figure 5.
Figure 5.
Graphical display of correlation between log ß1ADR AB and left ventricular ejection fraction
 Figure 6.
Figure 6.
Graphical display of correlation between log ß1ADR AB left ventricular end diastolic diameter
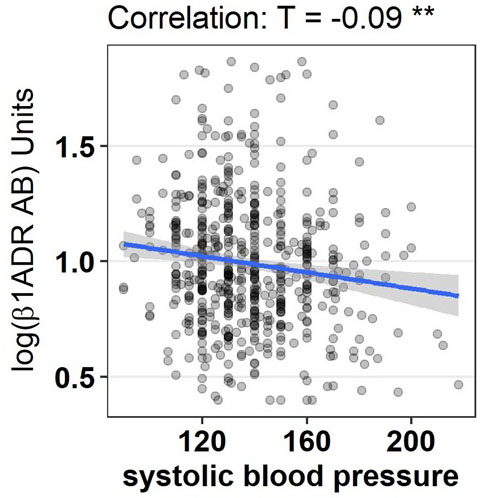 Figure 7.
Figure 7.
Graphical display of correlation between log ß1ADR AB and systolic blood pressure
In this paper we assess levels of ß1-ADR-AAs under titration of a cardio-selective vs. non-selective beta-blocker using a novel commercially available ELISA. To date, this is the largest assessment of ß1-ADR-AA in clinical heart failure patients. It should be noted that sensitivity and specificity of the assay employed has not yet been completely established. Nevertheless, this assay exhibits a clear association between ß1-ADR-AAs and cardiac dilatation as well as pump failure, which confirms to previous results obtained by others with functional assays currently considered a gold standard (20, 21, 22). We propose therefore that it can be considered as a valid surrogate marker for ß1-ADR-AA. ß1-ADR-AA were found to be higher in a setting of reduced LVEF, and higher in the setting of dilated ventricles. ß1-ADR-AA correlated positively with heart rate, but were not modified by the usage or dosage of beta-blockers.
Patients included in this analysis represent a typical heart failure population suffering from clinical HF irrespective of LVEF and of different aetiologies. In the CIBIS-ELD main trial, overall tolerability of titrating bisoprolol vs. carvedilol to target dose in elderly patients with heart failure was comparable. The pattern of intolerance, however, was different: bradycardia occurred more often in the bisoprolol group, whereas pulmonary adverse events occurred more often in the carvedilol group (23). The currently analysed study population represents a 65% share of the CIBIS-ELD trial patients. Patient distribution in the current sample was comparable to the main trial in regard key parameters, e.g. sex, age, NYHA class, BMI, blood pressure, heart rate, LVEF, and co-morbidities (23).
Antibodies specific for the ß1-ADR are found in patients with HF of various aetiologies (25). The mechanisms causing the autoimmune response in predisposed individuals are complex and not fully understood (26). Although the role of certain AA remains unclear, these antibodies actually contribute to the pathogenesis of HF (4). The epitopes most frequently targeted by the group of ß1-ADR-AA are located in the first and second extracellular loops of the ß1-ADR (27), (28). In regard to therapeutic options, immuno-adsorption (14), (15) and cardio-selective beta-blockade (16) have been investigated in this setting for more than a decade. Novel approaches, such as aptameres (5) and cyclic peptides such as the COR-1 peptide (18) and β1EC2-CP (29) are currently being debated.
Interestingly, the previously described association between ß1-ADR-AA and cardiac dilatation as well as pump failure was confirmed in the present heart failure population. In addition to previous work (10), (11), where altered levels of ß1-ADR-AA were restricted to DCM/ICM, but also found in a broader population of HF with different etiologies. Holthoff et al have described b1-ADR-AA in 60% of patients with dilatative cardiomyopathy, 8% of healthy volunteers and 17% of patients with hypertensive heart disease (21). Our findings may indicate a role of auto-immune response that is not restricted to DCM alone, hence in line previous works on Chagas’ cardiomyopathy (6) or ischemic heart disease (30), but not with others (11),
Resting heat rate has been identified a relevant risk factor and promising treatment target in patients suffering from heart failure (31), (32). Hence, the correlation observed between baseline ß1-ADR-AA and resting heart rate at baseline, at follow up 12 weeks later as well as in regard to change in HR during the study period (r=0.0.99, p=0.0.45) may bear significant clinical consequences. Interestingly, when looking at the change in HR and a change in ß1-ADR-AA between baseline and follow-up, this correlation was lost. This may partially be explained by the presence of a cocktail of ß1-ADR-AA with different functional (agonist vs. antagonist) entities involved in our observation, of which at least part of the detected AA are agonistic in function. Similar cocktails have recently described by Stavrakis et al (JACCS 54, 1309, 2009) using functional assays.
While we did observe an increase in ß1-ADR-AA levels under titration over time, we did not observe a relevant effect of the respective study medication titration on ß1-ADR-AA levels. Although treatment in this study differed in regard to a ß1-cardioselective beta-blockade with bisoprolol vs. a non-selective blockade with carvedilol, we assume that both compounds addressed the ß1-ADR similarly. This is in line with the main findings of the CIBIS-ELD trial (23). Interestingly, receptor-distal mechanisms have been discussed to enhance myocardial performance despite ongoing ß1-ADR antagonist occupancy (33). Bisoprolol (34) and carvedilol (35) have been used in animals to reduce G protein-coupled receptor kinase-2 expression. Jahns et al have used bisoprolol to interfere with the adenylyl cyclase response to ß1-ADR-AA (13). In fact, beta-blocker therapy may not be sufficient to ameliorate the ß1-ADR-AA induced end-organ damage. Novel double-acting therapeutic strategies that scavenges harmful anti-β1EC2-antibodies and also selectively deplete memory B-cells involved in the production of such antibodies by immuno-modulating cyclo-peptides alone or as an add-on to β1-blockade may represent a promising new therapeutic option in such immune-mediated heart failure (29).
The presently used and commercially available ELISA used to detect AA targeted against the cardiac ß1-ADR allowed for routine quantification of ß1-ADR-AA levels. Nikolaev et al (20) have previously developed a rapid method for the detection of activating autoantibodies directed against the ß1-ADR in HF. Their approach by also measuring ß1-ADR-mediated increase in intracellular cAMP by fluorescence resonance energy transfer (FRET) using a highly sensitive cAMP sensor prove to be fast and highly sensitive (20). In the presently used ELISA, it remains unclear if a conformational change in the g-protein-coupled receptor also results in an increased production of cAMP. Hence, some claim studies on g-protein-coupled receptor AA in cardiovascular disease inconclusive and misleading unless corroborated by measurements of IgG interactions with the respective native membrane-bound receptor(s) and/or by an assessment of their impact on receptor function, which includes their effects on receptor-mediated signalling (36, 37). Refining this approach, data from native cell-based competition ELISA (21) is suggestive of an AA agonist-like modulation of the ß1-ADR conformation (38). Aptameres (5) and cyclic peptides such as the COR-1 peptide (18) have been discussed (4) to optimize the solid phase of ELISA such as the ELISA routinely used. Interestingly, Holthoff’s novel whole-cell-based competition ELISA yielded a markedly lower rate of false-positives compared with peptide-based assays (21).
The currently used ELISA does not address whether the detected AA, act in agonist-like, neutral or antagonist-like functions. Possibly, the presently used ELISA quantifies a cocktail of ß1-ADR-AA with different functional (agonist vs. antagonist) or non-functional entities. It should be noted that sensitivity and specificity of the assay employed has not yet been completely established. Nevertheless, the values for ß1-ADR-AAs measured with this assay exhibit a clear-cut association with cardiac dilatation as well as pump failure, which conforms to previous results obtained by others with functional assays currently considered a gold standard. We propose therefore that these results can be considered as a clinically valid surrogate marker for ß1-ADR-AA. Our data show clinical effects which may partially be explained by ß1-ADR-AA of which at least part of are agonistic in function. Future studies will have to clarify this important question. To further prospectively address cardiac autoimmunity and the specific cardio-noxious role ß1-ADR-AA bear as disease-modifier or disease-triggers, Jahns et al currently conduct a trial on aetiology, titre-course, and survival (ETiCS) (22) in HF patients.
Although data and blood samples of the CIBIS-ELD trial were prospectively collected, the character of the present secondary post-hoc analysis is a hypothesis generating description and warrants further clarification. While the used ELISA can quantify the ß1-ADR-AA serum levels, the quality of the AA; i.e. agonist-like or antagonist-like functions, remains unclear.
This novel ELISA, utilizing ß1-ADR in its physiological condition, offers the possibility to measure ß1-ADR-AA in clinical routine. The ELISA allowed for pathophysiological insights: ß1-ADR-AA levels significantly increased under titration with beta-blockers (p<0.0.1), irrespective of type of BB. Higher levels of ß1-ADR-AA at baseline are associated with higher heart rates, lower ejection fraction and enlarged left ventricles, indicating a role of these antibodies in the heart failure.
The specificity of the new ELISA has to be further investigated to elucidate the quality of detected antibodies in respect to their agonist vs. antagonist like functions. Studies assessing Etiology, Titre-Course, and effect on Survival (ETiCS) of beta1-adrenoceptor antibodies are needed to further elucidate the described phenomenon.
Hans-Dirk Dungen and Ralf Dechend are senior authors. We wish to express our gratitude towards all patients and research personnel for their participation, support and critical revision of this analysis, especially Svetlana Apostolovic, Goran Loncar, Simone Inkrot, Agnieszka Topper, Max Fritschka, Julia Schönleber, Rolf Wachter, Verena Tscholl, Mitja Lainscak. This work was supported by the Competence Network Heart Failure, funded by the Federal Ministry of Education and Research (BMBF), FKZ 01GI0205. The CIBIS-ELD trial was supported by an unrestricted research grant from Merck KGaA, which had no influence on the trial conduct, results, analysis, publication or any other claim to intellectual property rights resulting from the trial. Blood analysis was performed by CELLTREND (Luckenwalde, Germany) free of charge. TDT, DNM, DO, FE, ET, NW, BP, HDD, RD have nothing to disclose. GR and DD have contributed to the development of the used ELISA. AH has received fees for statistical analysis. HH is CEO and stakeholder of Celltrend GmbH.