







Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.
 , Guangzhou Xu 1
, Guangzhou Xu 11 Department of Oral Surgery, Shanghai Ninth People's Hospital affiliated to Shanghai Jiaotong University, School of Medicine, Shanghai Key Laboratory of Stomatology, Shanghai, P. R. China
2 Department of Pediatric Dentistry, Shanghai Ninth People's Hospital affiliated to Shanghai Jiaotong University, School of Medicine, Shanghai Key Laboratory of Stomatology, Shanghai, P. R. China
3 Department of Oral and Maxillofacial-Head and Neck Oncology, Shanghai Ninth People's Hospital affiliated to Shanghai Jiaotong University, School of Medicine, Shanghai Key Laboratory of Stomatology, Shanghai, P. R. China
Abstract
miR-139-5p has a tumor suppressor effect in some cancers and negatively regulates CXCR4. To this end, we examined the expression and mechanism of of action of miR-139-5p and CXCR4 in oral squamous cell carcinoma (OSCC). miRNA-139-5p was down-regulated whereas CXCR4 was increased in tissues and cells of OSCC. Moreover, low expression of miR-139-5p was associated with a low survival. Overexpression of miR-139-5p in OSCC inhibited in vitro and in vivo cell proliferation and in vitro mobility of OSCC and inhibited the expression of WNT responsive c-myc, cyclinD1, and Bcl-2, and such effects were all reversible by an inhibitor of miR-139-5p or over-expression of CXCR4. The inverse relation between expression of miR-139-5p and CXCR4 might be related to the fact that miR-139-5p negatively regulates CXCR4 expression by virtue of direct binding. These findings underscore the importance of miR-139-5p and CXCR4 in regulation of OSCC.
Keywords
- Oral
- Squamous Cell Carcinoma
- miR-139-5p
- Wnt
- Beta-Catenin Signaling
- Proliferation
- Invasion
Oral squamous cell carcinoma (OSCC) accounts for approximately 3% of all newly diagnosed cancers and is one of the most commonly diagnosed head and neck cancers that carries a poor prognosis and a low survival rate (1). Despite the significant progress over the past several decades in undestanding the signals that cause tumor progression, the overall 5-year and disease-free survival rates remain, respectively, at 61% and 75% in young patients (2). Therefore, it is essential to elucidate the underlying molecular mechanisms that drive OSCC.
microRNAs (miRNAs) are a class of non-coding RNA molecules, made of 20-22 nucleotides. miRNAs are vital to multiple biological processes, and their key roles, in the context of tumor development, are beginning to be appreciated. Human cancers show dys-regulated expression of multiple miRNAs, that confer the malignant cells their tumorigenic potential. miRNAs regulate gene expression by virtue of degradation and post-transcriptional repression by direct binding to the 3’ untranslated region (3’-UTR) of the complementary mRNA sequences (3-4). Each miRNA regulates the expression of hundreds of messenger RNAs (mRNAs) and each mRNA can be regulated by a host of diverse miRNAs (5). Emerging evidence has shown that the initiation, development, and progression of malignancy including OSCC might be disrupted by diverse miRNAs including miR-133a-3p, miR-26a and miR-200c (5-9). Consistent with such a role, overexpression of miR-133a-3p or miR-200c in OSCC cells suppressed their proliferation (7-8). In contradistinction to this subset of miRNAs, overexpression of miR-654-5p promotes the proliferation, metastasis, and chemoresistance of OSCC (10).
MiR-139-5p has been described in a limited number of studies as a potential tumor suppressor miRNA and a biomarker for tumor diagnosis (11-14). However, little is known about the function(s) of miR-139-5p in the OSCC. CXC chemokine receptor 4 (CXCR4) is a G protein-coupled receptor (GPCR) with diverse functional roles including angiogenesis, hematopoiesis, neurogenesis and immune function (15-16). Recently, it was proposed that miR-139-5p negatively regulates CXCR4 during endothelial maturation, and vascular maturation (16-17). Such an effect is important, since it was shown that pharmacological inhibition of CXCR4 signalling or augmentation of the miR-139-5p-CXCR4 axis ameliorated the vascular phenotype of APLN/APLNR deficient state (18). This crosstalk involves miR-139-5p, which directly targets CXCR4 in endothelial cells (18). Here, we characterize the participation of miR-139-5p in OSCC with respect to cell proliferation and mobility which are cardinal features of malignant cells. In addition, we examined how the expression of miR-139-5p in OSCC correlated with the expression of CXCR4. Moreover, by modifying the expression of miR-139-5p and CXCR4, we examined how this interplay impacts the tumor cell proliferation in vitro and in vivo and cell mobility in vitro.
Proteins were quantified by a bicinchoninic Acid Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Molecular weight markers were purchased from ThermoFisher (Waltham, MA, USA). For Western blotting, we used antibodies to CXCR4 (ThermoFisher, PA3-305), beta-actin (ThermoFisher, MA5-15739), beta-catenin (ThermoFisher, 71-2700), c-myc (ThermoFisher, MA1-980), Cyclin D1 (ThermoFisher, MA1-39546), and to Bcl-2 (ThermoFisher, PA1-37161). Normal mouse IgG and human anti-(Ago2) antibody was obtained from Millipore (Bedford, MA, USA). Trizol reagent was purchased from Invitrogen (Carlsbad, CA, USA). Quantitative real-time PCR (qRT-PCR) was carried out by using QuantStudio™ 3 Real-Time PCR Systems (ThermoFisher, USA). For transient in vitro transfection, we used lipofectamine (Invitrogen, Carlsbad, CA, USA) and for in vivo transfection we used Invivofectamine 2.0 (Invitrogen, Carlsbad, CA, USA). Magna RIP kit was purchased from Millipore (Bedford, MA, USA).
The clinical studies in this report were approved by the institutional ethics committee. After obtaining consents from patients, sample of oral squamous cell carcinoma and adjacent non-tumorous tissues were obtained from 36 patients that did not receive chemotherapy or radiotherapy before surgery.
All animal experiments were approved by the Institutional Animal Care and Use Committee. Female BALB/c null 4-6-week-old mice were purchased from Vital River (Beijing, China).
Normal, spontaneously immortalized, human oral keratinocytes (hNOK), human oral squamous cell carcinoma cell lines (KON, SAS) were obtained from American Type Culture Collection of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). Cells were maintained in Dulbecco’s Modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and antibiotics (100 µg/ml streptomycin and 100 U/ml penicillin). All cells were incubated in a humidified incubator containing 5% carbon dioxide at 37°C. The medium was changed every two days.
KON or SAS Cells were seeded in 6-well culture plates at 2 × 106 cells per well. When reaching 70%-80% confluence, cultured cells were transfected using lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) along with the miR-139-5p mimics, miR-139-5p inhibitor, si-CXCR4, pcDNA-CXCR4 and their corresponding control (miR-NC, in-miR-NC, si-NC, pcDNA) according to the manufacturer’s instructions. Mock cells received only transfection agent. Cells (5×103) were cultured in 6-well plates for 48 hours, and then cells were removed and plated in 96 well plates for 24, 48, 72 and, 96 hours post-transfection and the transfection efficiency was assessed by quantitative real-time polymerase chain reaction (qRT-PCR) or Western blotting. The sequences used in this study are listed in Table 1.
| miRNA and siRNA and control | Sequence 5'- 3' |
|---|---|
| miR-139-5p mimics | UCUACAGUGCACGUGUCUCCAGU |
| miR-NC | UUCUCCGAACGUGUCACGUTT |
| miR-139-5p inhibitor | ACUGGAGACACGUGCACUGUAGA |
| in-miR-NC | CAGUACUUUUGUGUAGUACAA |
| si-CXCR4 | CCGACCUCCUCUUUGUCAUTT |
| si-NC | UUCUCCGAACGUGUCACGUTT |
| CXCR4 | Accession:HQ537555 |
Cell proliferation was assessed by adding 20 µl of MTT (5 mg/ml) and cultures were incubated at 37°C. Following a 2 hr, 150 µl of dimethyl sulfoxide (DMSO) was added to lyse formazan crystals and cells were incubated at room temperature for 10 minutes. Then the absorbance was measured at 490 nm for each well using a microtiter plate reader (BioTek ELX800, Winooski, VT, USA).
Mobility of cells was assessed by using a transwell chamber coated with matrigel. Cells (2×105) were suspended in a 150 μL serum-free medium and these were added to the upper chamber of transwell chambers. 500 μL of DMEM medium with 10% FBS was added to the lower chambers. After culturing for 24 h, cells on the top surface of the inserts were removed. Cells that migrated into the lower side of the membranes were fixed with 4% PFA (paraformaldehyde) and stained with 0.1% crystal violet. The number of cells was counted under immersion oil using a light microscope. The results were expressed as the number of cells that passed the membranes to the total number of cells added to each chamber.
Cells were lyzed in RIPA lysis buffer. Proteins in cell lysates were quantified by a Bicinchonnic Acid Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Proteins (50 μg) were resolved in SDS-PAGE gels for 60 minutes at 120 volt, and they were then transferred at 55V for 4 h at 4°C to polyvinylidene difluoride (PVDF) membranes. After blocking with 5% non-fat dry milk in TBS, membranes were incubated with primary antibodies: polyclonal rabbit anti-CXCR4 (1:200; PA3-305, ThermoFisher, Waltham, MA, USA), polyclonal rabbit anti-beta-catenin (1:200; 71-2700, ThermoFisher, Waltham, MA, USA), monoclonal mouse anti-c-myc (1:500; MA1-980, ThermoFisher, Waltham, MA, USA), recombinant monoclonal rabbit anti-CyclinD1 (1:500; MA1-39546, ThermoFisher, Waltham, MA, USA), rabbit anti-Bcl-2(1:50; PA1-37161, ThermoFisher, Waltham, MA, USA), and Mouse anti-beta-actin (1:5000; MA1-140, ThermoFisher, Waltham, MA, USA) in TBS overnight at 4°C. Then, the membranes were washed three times for 10 minutes in TBS-T. Membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies: goat-anti-rabbit (ab205178, 1:10000, Abcam, Cambridge, MA, USA) and goat-anti-mouse (A16017; 1:5000, ThermoFisher, Waltham, MA, USA) for 1 h at room temperature. The immunoreactive proteins were revealed using the ECL system (Pierce Biotechnology, Rockford, IL, USA), and analyzed with Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Total RNAs were extracted by using Trizol reagent according to the manufacturer’s protocol. QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA, USA) was used to reverse transcribe the cDNA from mRNA. MiScript II RT Kit (Qiagen, Valencia, CA, USA) was used to transcribe the miR-139-5p. Then, qRT-PCR was carried out using SYBR Green (Promega, Madison, WI, USA) and QuantStudio™ 3 (ThermoFisher, Waltham, MA, USA). U6 small nuclear RNA (snRNA) was used to normalize miR-139-5 and GAPDH was used to normalize CXCR4. The relative expression of RNA was calculated by the 2-ΔΔCt method. Primer sequences used in this study are listed in Table 2.
| Gene | Forward 5'- 3' | Reverse 5'- 3' |
|---|---|---|
| GAPDH | GTCAACGGATTTGGTCTGTATT | AGTTTCTGGGTGGAGTGAT |
| U6 | GTTGACATCCGTAAAGACC | GGAGCCAGGGCAGTAA |
| CXCR4 | GAAGTGGGGTCTGGAGACTAT | TTGCCGACTATGCCAGTCAAG |
| miR-139-5p | GCCTCTACAGTGCACGTGTCTC | CGCTGTTCTCATCTGTCTCGC |
The KON or SAS cells were transfected with miR-139-5p or miR-NC (negative control). After 48 hours, cells were lysed in RIP lysis buffer containing a protease inhibitor cocktail and an RNase inhibitor followed by RIP assay using the Magna RIP kit according to the manufacturer’s protocol. In brief, the cell lysates were incubated with magnetic beads conjugated with human anti-(Ago2) antibody or normal mouse IgG (negative control). Then, proteins were digested with 1 μL 20 mg/mL proteinase K for 1 h, and the immunoprecipitated RNAs were isolated and purified by using Trizol reagent according to the manufacturer’s protocol. Then, to identify the binding targets, immunoprecipitated RNAs were then subjected to qRT-PCR.
The wild-type 3’UTR of CXCR4 and the mutant form (ACUGUAG to UGACAUC) were synthesized by Gemma pharmaceutical technology co., LTD. (Shanghai, China) and cloned into the pmirGLO dual-luciferase vector (Promega, Madison, WI, USA). Then, the vectors and miR-139-5p or negative control were co-transfected into KON and SAS cells using Lipofectamine 3000 according to the protocol (Invitrogen, Carlsbad, CA, USA). 48 hours after transfection, the luciferase activity was measured with a dual-luciferase report assay system (Promega, Madison, WI, USA) and results were normalized to the firefly luciferase activity.
To establish the xenograft tumor model, 2x106 KON cells were subcutaneously inoculated into the upper flank of the nude mice. The size of tumors was measured every 7 days. Animals were sacrificed at the end of the experiment, and tumors were excised, weighed and immediately flash frozen in liquid nitrogen before being stored at –800C. Subsequently, the frozen tumor tissues were utilized for qRT-PCR and Western blot assays to examine the expression levels of miR-139-5p, CXCR4, beta-catenin, c-myc, CyclinD1 and Bcl-2, respectively. Xenograft tumor tissues were weighed and immediately flash frozen in liquid nitrogen before being stored at –800C. Subsequently, the frozen tumor tissues were utilized for qRT-PCR and Western blot assays.
Data were analyzed with GraphPad Prism 7.0 software and expressed as mean ± standard deviation (S.D.). Significant differences between different groups were analyzed by one-way ANOVA or student’s t-test. All in vitro experiments were performed at least in triplicates. Differences between groups were considered to be statistically significant when P<0.05.
We characterized the expression of miR-139-5p by qRT-PCR in 36 OSCC and adjacent non-tumorous tissues. Compared to non-involved tissues, the expression of miR-139-5p was lower in majority of OSCC (Figure 1A). Consistent with this, the expression of miR-139-5p was significantly higher in human normal oral keratinocytes cell line, hNOK as comaped to the expression of this miRNA in OSCC cell lines, KON and SAS (Figure 1B). To further investigate OSCC tumor samples, the tumor samples were divided into low group (n=23) and high group (n=13) by the median expression of miR-139-5p. More importantly, the expression of miR-139-5p in patients with OSCC was lower in patients with a poor survival as compared to those with s longer survival (Figure 1C).
To decipher how the miR-139-5p regulates OSCC cell behavior, we over-expressed miR-139-5p in KON and SAS cells using the microRNA mimic. The success of transfection was assessed by the marked increase in expression of miR-139-5p in cells transfected miR-139-5p and not those that were mock transfected or were transfected with control miRNA (miR-NC) (Figure 2A). The miR-139-5p overexpression reduced proliferation of OSCC cells and not the hNOK cells (Figure 2B-D). Cells that were transfected with miR-139-5p showed a significantly lower mobility as compared to mock transected cells or cells that were transfected with miR-NC (Figure 2E).
To predict the downstream targets of miR-139-5p, we used the TargetScan Tool (http://www.targetscan.org). This analysis revealed CXCR4 as being post-transcriptionally repressed by miR-139-5p. To functionally test such repression, luciferase vectors were constructed that included either the partial sequence of wild or mutant 3’-UTR of CXCR4 (Figure 3A). These constructs were transfected into KON and SAS cells either with miR-139-5p mimic or negative control (blank control). The luciferase assay showed that luminescence intensity was significantly reduced when KON or SAS cells were transfected concomitantly with the wild-type 3’UTR of CXCR4 and miR-139-5p mimic, and not after transfection with the mutant sequence (Figure 3B and 3C). On the other hand, luciferase activity was significantly increased when KON, or SAS cells were co-transfected with the wild-type 3’-UTR of CXCR4 and the miR-139-5p inhibitor (Figure 3C). RIP assay showed direct binding of miR-139-5p to the 3’-UTR of CXCR4 (Figure 3D and 3E). Overexpression of miR-139-3p induced higher expression level of CXCR4 mRNA than transfection with miR-NC in Ago2 RIP group, however, there was little enrichment efficacy in the IgG RIP group (Figure 3F). Moreover, Western blot assay suggested that the expression level of CXCR4 was markedly inhibited in KON and SAS cells transfected with miR-139-3p when compared with cells transfected with miR-NC (Figure 3G). The overexpression of miR-139-3p led to decreased levels of beta-catenin, c-myc, CyclinD1 and Bcl2, implying that miR-139-3p upregulation could inhibit the Wnt/β-catenin pathway in OSCC cells (Figure 3H and 3I).
Given that miR-139-5p negatively regulates CXCR4, we considered to test whether such down-regulation can suppress pathways that CXCR4 regulates. Previously, it was shown that downregulation of the CXCR4/CXCL12 axis blocks the activation of the Wnt/β-catenin pathway in human colon cancer cells (19). First, Western blot assay suggested that miR-139-5p downregulation overturned the inhibitory effect of si-CXCR4 on CXCR4 level in KON and SAS cells (Figure 4A). To show that the WNT members are jointly regulated by miR-139-5p and CXCR4, we first down-regulated CXCR4 by siRNA and then examined the total protein levels of beta-catenin, c-myc, CyclinD1 and Bcl2. This analysis showed that the level of WNT protein members were reduced significantly in KON and SAS cells showing that CXCR4 actively regulates WNT members (Figure 4A and 4B). To show regulation by miR-139-5p, we inhibited it by miR-139-5p inhibitor. This inhibition in cells with silenced CXCR4 led to a significant increase in beta catenin members (Figure 4B and 4C). In addition, western blot results suggested that the overexpression of miR-139-5p could block the protein level of CXCR4 in in KON and SAS cells (Figure 4D). To further verify the effect of miR-139-5p on the levels of beta-catenin, c-myc, CyclinD1 and Bcl2, we over-expressed the level of miR-139-5p in KON and SAS cells. As shown in Figure 4E and 4F, the level of beta-catenin, c-myc, CyclinD1 and Bcl2 were reduced by 80%, 75%, 80%, 60% in KON, and 85%, 65%; 70%, 75% in SAS cells due to the overexpression of miR-139-5p compared to mock or negative control transfected cells.
Given that miR-139-5p directly binds the 3’-UTR of CXCR4 and it negatively regulate CXCR4 expression, we considered that such an inverse relationship might also exist in OSCC and that such an axis operates to regulate tumor cell proliferation and mobility. Consistent with such a prediction, the CXCR4 expression both at the mRNA and protein level was as high as 2.1 fold in OSCC tumor tissues (n=36) as than that was observed in the adjacent non-tumorous tissues (n=36) (Figure 5A-5B). These analysis showed that the expression of miR-139-5p negatively and significantly correlated with CXCR4 expression in OSCC (Figure 5C). These results showed that CXCR4 was a downstream target mRNA of miR-139-5p and negatively correlated with miR-139-5p in OSCC.
To better understand the relationship between miR-139-5p and CXCR4, we over-expressed both miR-139-5p and CXCR4 in OSCC cells. The expression of CXCR4 was decreased when miR-139-5p was overexpressed in KON and SAS cells, this effect was reversed by co-transfection of cells with pcDNA-CXCR4 (Figure 5D).To determine the existense of miR-139-5p/CXCR4 axis, we transfected in KON and SAS cells with miR-139-5p without and with CXCR4 and then we assessed the proliferation and mobility of tumor cells. These experiments showed that CXCR4 reverses the inhibitory effects of miR-139-5p in cell proliferation and mobility of tumor cells (Figure 5E-5G).
To investigate whether miR-139-5p could inhibit the tumor growth in vivo, we injected KON cells to 15 female BALB/c null 4-6-week nude mice that were grouped into three groups (each group n=5) that received mock, miR-NC, or miR-139-5p, by injecting different oligos in a solution containing 0.1 mL Invivofectamine 2.0 around the tumor once a week for 5 cycles. Tumor volumes were examined once a week. Five weeks later, tumors were carefully excised, weighed. This study showed that tumor size and weight dropped in presence of miR-139-5p upregulation, suggesting that stable overexpression of miR-139-5p efficiently suppressed cell growth of HCC in vivo (Figure 6A and 6B).
The analysis showed miR-139-5p was overexpressed in the tumors of animals that received miR-139-5p and not mock or miR-NC group (Figure 6C). Western blot analysis also showed the level of CXCR4 and WNT members were lower in the tumors of animals that received miR-139-5p and not mock or miR-NC group (Figure 6D-6E).
Recently, increasing studies have demonstrated that miRNAs can act as either oncogenes or tumor suppressors, both in vitro and in vivo (20). These miRNAs are involved in the proliferation, migration, invasion, apoptosis, angiogenesis, and epithelial-mesenchymal transition (EMT) (21). miR-139-5p was reported to be a tumor suppressor in different cancers, including bladder cancer (22), myeloid leukemia (23), gastric cancer (24), endometrial cancer (25) and osteosarcoma (26). In the present study, it was shown that miR-139-5p was down-re-gulated in OSCC tissues and cell lines. In addition, this low-expression of miR-139-5p correlated with a poor survival in OSCC patients. We also show that consistent with the idea that miR-139-5p is a tumor suppressor, overexpression of miR-139-5p in OSCC cell line KON and SAS regulated proliferation and mobility of the tumor cells in vitro and restrained tumor growth in vivo. There are other indications the role of miR-139-5p as a tumor suppressor by shown in different tumors that miR-139-5p regulates proliferation, mobility and invasive property of the tumor cells by a diverse group of targets.
It appears that miR-139-5p acts as a tumor suppressor by a diverse group of targets. For example, in colorectal cancer, miR-139-5p, by virtue of downregulation of AMFR and NOTCH1, inhibited the migration of tumor cells (13). MiR-139-5p also suppressed the migration of tumor cells by targeting ZEB1 and ZEB2 in hepatocellular carcinoma cells (18). MiR-139-5p was also shown to inhibit the EMT and enhanced the chemosensitivity of colorectal cancer via binding to BCL2 (27). It was shown that in uterine leiomyoma, miR-139-5p regulates proliferation, apoptosis, and cell cycle via targeting TPD52 (28). In bladder cancer, miR-139-5p inhibits proliferation by targeting the BMI1 oncogene (29).
By using bioinformatical analysis in OCSS, we identified CXCR4 as a putative target of miR-139-5p. The validity of such prediction that CXCR4 is a downstream mRNA was then substantiated by luciferase and RIP assays. These studies clearly demonstrated CXCR4 to be transcriptionally suppressed by miR-139-5p. We further validated this by showing that overexpression miR-139-5p led to suppression of CXCR4 and that CXCR4 overexpression reversed the inhibitory effects of miR-139-5p on cell proliferation and mobility of OSCC. Taken together, miR-139-5p inhibits OSCC progression via directly regulating CXCR4 and validates the role of miR-139-5p as an active tumor suppressor. CXCR4, belongs to the superfamily of seven-transmembrane G-protein-coupled receptors, is expressed in many cell types and even in cancer cells (30). CXCR4 and its ligand CXCL12 appear to be involved in tumor progression, metastasis, development, and survival (31). In addition, CXCR4 was shown to activate the Wnt/beta-catenin pathway (32), which has been implicated in tumorigenesis of various cancers (33).
In conclusion, this study suggests that miR-139-5p plays an inhibitory role in OSCC development and progression via directly regulating the target gene, CXCR4 and its down-stream Wnt/beta-catenin signaling pathway in OSCC (Figure 7).
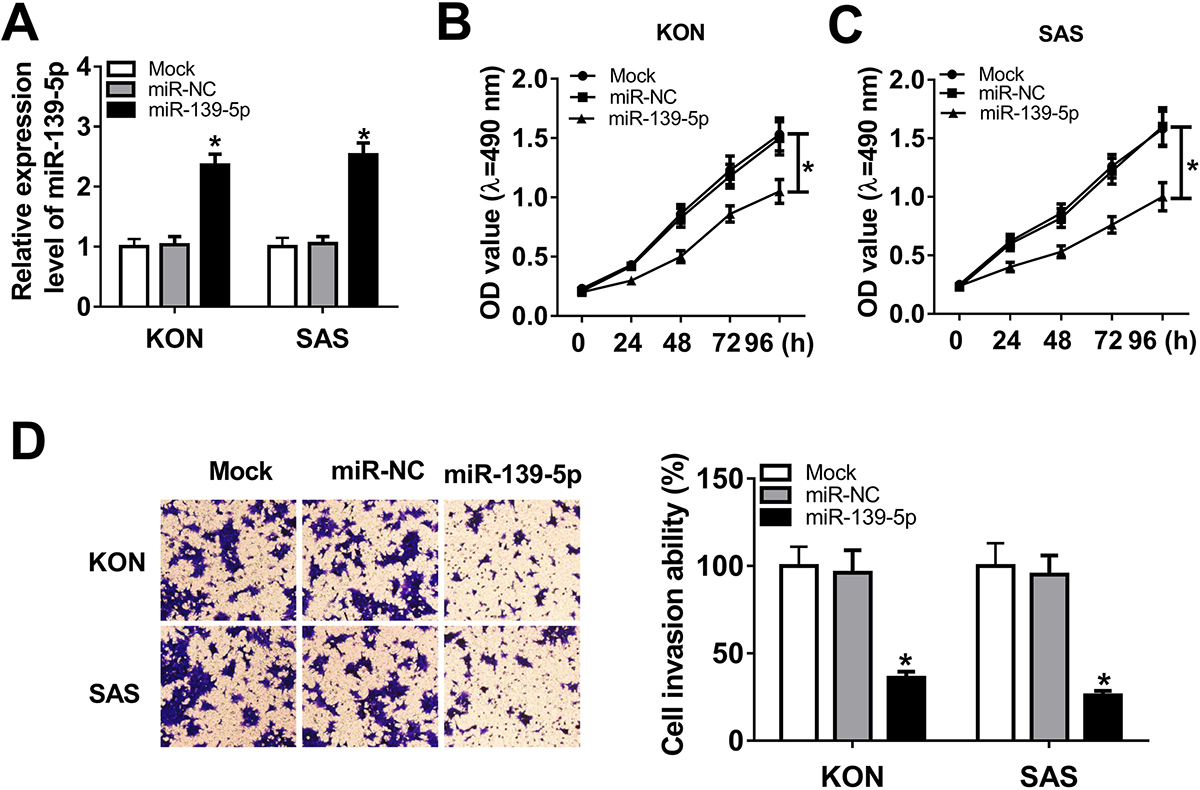 Figure 2
Figure 2miR-139-5p overexpression inhibited cell proliferation and invasion in OSCC. (A) miR-139-5p expression in KON or SAS cells transfected with mock, miR-NC or miR-139-5p, individually. The relative expression of miR-139-5p in KON cells was 1±0.13 in mock transfected, 1.03±0.14 in those transfected with control miRNA (miR-NC) and 2.36±0.18 in cells that were transfected with miR-139-5p. The relative expression of miR-139-5p in SAS cells was 1±0.15 in mock transfected cells, 1.05±0.12 in cells transfected with miR-NC and 2.53±0.20 in those that were transfected with miR-139-5p. The proliferation of KON (B), SAS (C) and hNOK (D) cells transfected with mock, miR-NC and miR-139-5p at 0, 24, 48, 72, and 96 hours was assessed with MTT assay. (E) Transwell assay was used to assess the mobility of KON or SAS cells after mock transfection or transfection with miR-NC or miR-139-5p. The percent of cells that traversed the membranes were 100±11% in mock, 96±13% in miR-NC and 36±3.6% in miR-139-5p transfected cells. Data are expressed as means ± S.D. *p < 0.05.
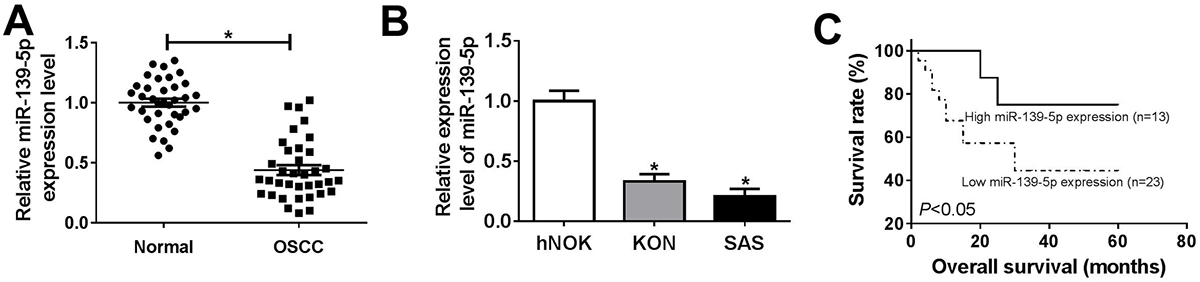 Figure 1
Figure 1miR-139-5p was downregulated in OSCC and related to survival. Based on the median value of miR-139-5p level, in 36 OSCC the tumors were classified into those with a high miR-139-5p (n=13) and low expression groupgroup (n=23). (A) The miR-139-5p expression in normal (n=36) and OSCC tissues (n=36). (B) The expression of miR-139-5p in human normal oral keratinocytes cell line hNOK, human oral squamous cell carcinoma cell line KON and SAS. (C) The tumor samples were divided into low group (n=23) and high group (n=13) by the median expression of miR-139-5p, and then the correlation between the overall survival of OSCC patients and miR-139-5 level was determined by Kaplan-Meier analysis. Data are expressed as means ± S.D. *p < 0.05. OSCC: oral squamous cell carcinoma.
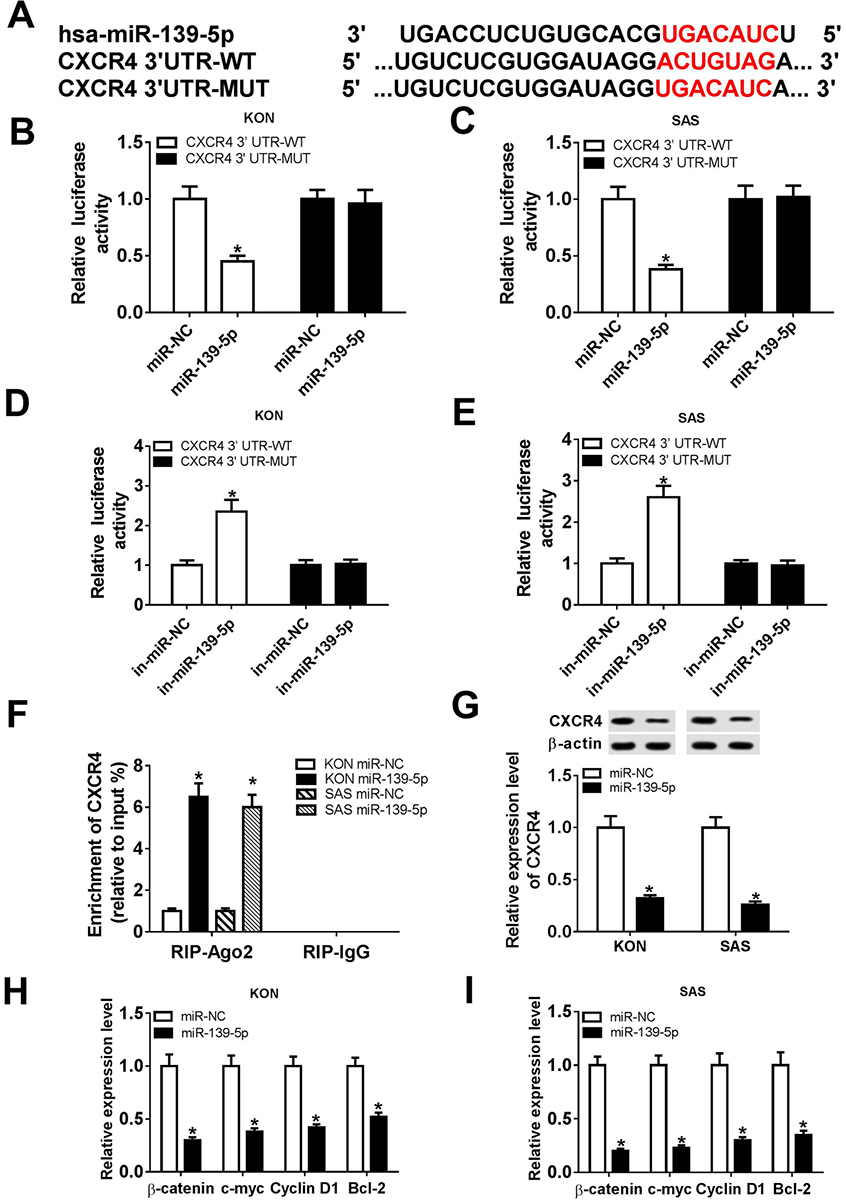 Figure 3
Figure 3CXCR4 was a target of miR-139-5p. The binding sites of miR-139-5p and CXCR4 were predicted by TargetScan Tool. (B and C) Relative luciferase activity of KON and SAS cells co-transfected with miR-NC or miR-139-5p and CXCR4 3’-UTR-WT or CXCR4 3’-UTR-MUT. (D and E) Relative luciferase activity of KON and SAS cells co-transfected with in-miR-NC or in-miR-139-5p and CXCR4 3’-UTR-WT or CXCR4 3’-UTR-MUT. (F) RIP assay was used to confirm the relationship between miR-139-5p and CXCR4. (G) The protein level of CXCR4 in KON and SAS cells was detected. (H and I) The mRNA levels of beta-catenin, c-myc, CyclinD1, and Bcl-2 were detected in miR-139-5p-transfected in KON and SAS cells. Data are expressed as mean ± S.D. *P < 0.05, r=-0.7735, miR-NC: negative control. in-miR-NC: negative control inhibitor. in-miR-139-5p: miR-139-5p inhibitor.
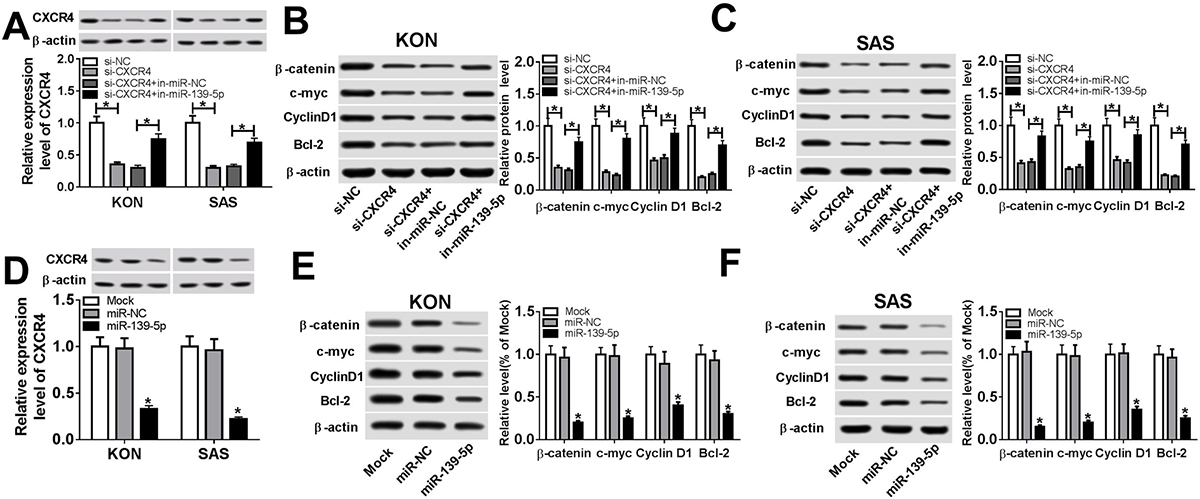 Figure 4
Figure 4miR-139-5p regulated Wnt/beta-catenin pathway via CXCR4. (A) Protein expression of CXCR4 in miR-139-5p inhibitor and si-CXCR4-transfected KON or SAS cells. (B) Protein expression of beta-catenin, c-myc, CyclinD1, and Bcl-2 in KON cells after transfection of miR-139-5p inhibitor and si-CXCR4. (C) Protein expression of beta-catenin, c-myc, CyclinD1, and Bcl-2 in SAS cells after transfection of miR-139-5p inhibitor and si-CXCR4. (D) Protein expression of CXCR4 in miR-139-5p overexpression-transfected KON or SAS cells. (E) Protein expression of beta-catenin, c-myc, CyclinD1, and Bcl-2 in KON cells after miR-139-5p overexpression. (F) Protein expression of beta-catenin, c-myc, CyclinD1, and Bcl-2 in SAS cells after miR-139-5p overexpression. Data are expressed as mean ± S.D. *p < 0.05. miR-NC: negative control. si-NC: siRNA of negative control. si-CXCR4: siRNA of CXCR4.
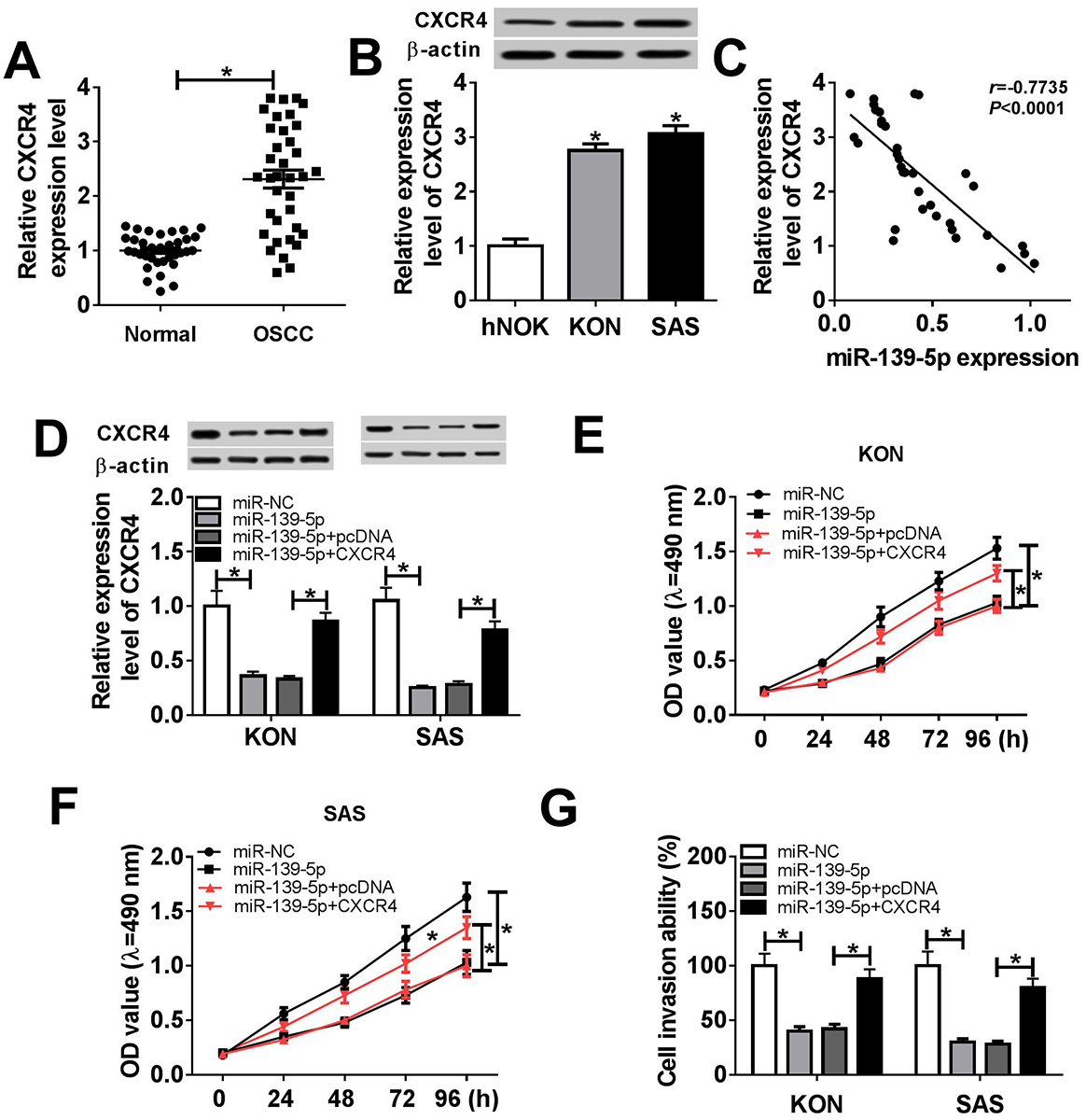 Figure 5
Figure 5CXCR4 restoration reversed the effects of miR-139-5p. (A) CXCR4 mRNA expression in normal (n=36) and OSCC (n=36) patients. (B) The protein level of CXCR4 in human normal oral keratinocytes cell line hNOK, human oral squamous cell carcinoma cell lines (KON and SAS). (C) The correlation between miR-139-5p and CXCR4 expression in OSCC patients. (D) CXCR4 protein expression in KON and SAS cells transfected with miR-NC, miR-139-5p, miR-139-5p + pcDNA, and miR-139-5p + CXCR4. (E) MTT assay was introduced to measure cell proliferation of KON cells transfected with miR-NC, miR-139-5p, miR-139-5p + pcDNA, and miR-139-5p + CXCR4. (F) The proliferation of SAS cells after the transfection with miR-NC, miR-139-5p, miR-139-5p + pcDNA, and miR-139-5p + CXCR4. (G) The cell invasion ability of KON and SAS cells transfected with miR-NC, miR-139-5p, miR-139-5p + pcDNA, and miR-139-5p + CXCR4. Data are expressed as mean ± S.D. * p < 0.05. miR-NC: negative control. CXCR4: pcDNA-CXCR4. pcDNA: negative control vector.
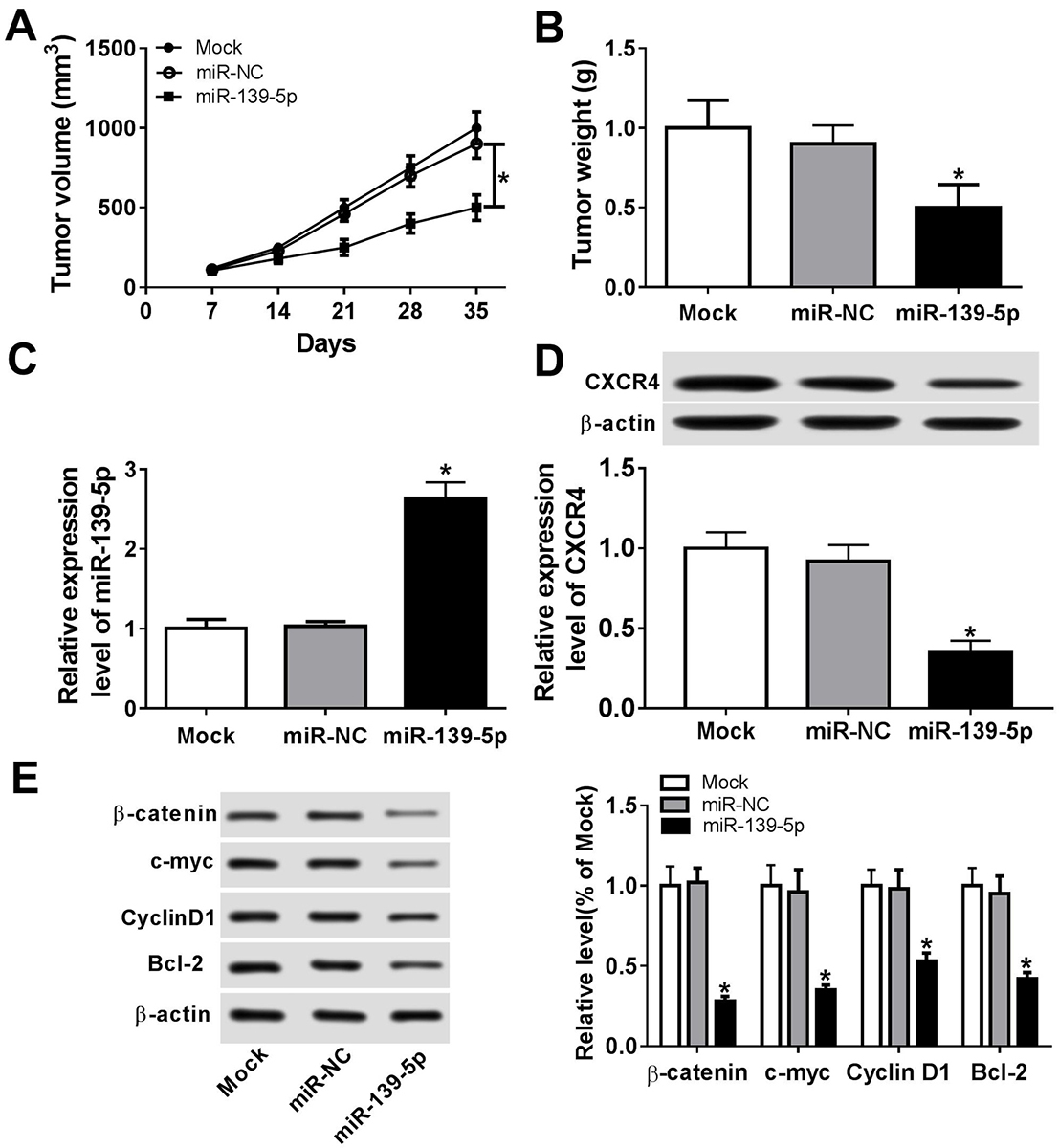 Figure 6
Figure 6miR-139-5p inhibited tumor development in vivo. 2x106 KON cells were subcutaneously inoculated in the upper flank of the nude mice. On day 7, 10 nmol miR-139-5p or miR-NC or no vector (Mock) was injected around the tumors with 0.1 mL Invivofectamine 2.0 (Invitrogen, USA) once per week per site for 4 weeks. Each group had 5 mice. (A) Tumor volume was measured once a week after injection. (B) The tumor weight in each group on day 35. (C) miR-139-5p expression in the tumor tissues of the nude mice on the 35th day. (D) CXCR4 expression in the tumor tissues on the 35th day. (E) The expression of beta-catenin, c-myc, CyclinD1, and Bcl-2 in tumor tissues on day 35. Data are expressed as mean ± S.D. *p < 0.05.
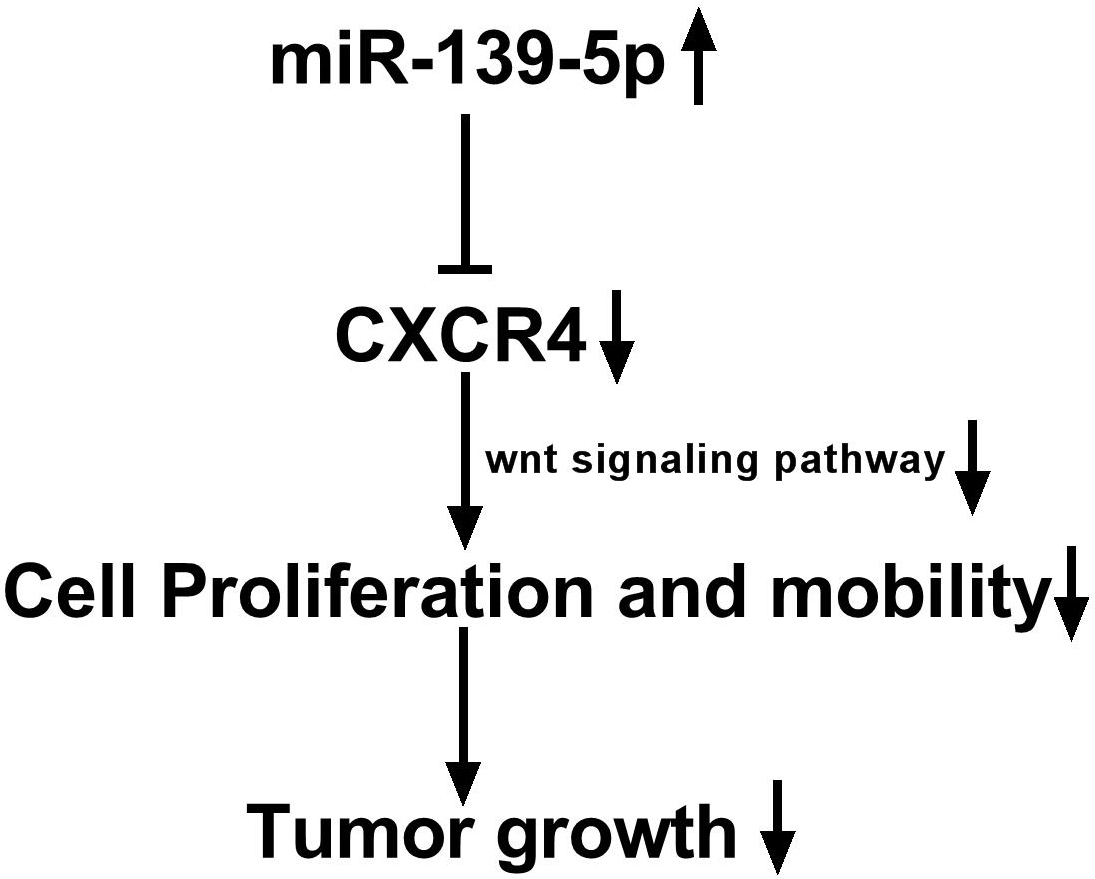 Figure 7
Figure 7miR-139-5p regulates the progression of OSCC by targeting CXCR4.
This work was supported by the Youth Science Fund Project of National Natural Science Foundation of China (Grant no. 31800806)
OSCC
Oral Squamous Cell Carcinoma
RNA Immunoprecipitation
3' Untranslated Region
Epithelial–Mesenchymal Transition