
Background: Mitochondrial dysfunction plays a crucial role in Parkinson’s disease (PD) pathogenesis. The present study was undertaken to investigate the effects of Telmisartan (TEL), an angiotensin II type 1 receptor (AT1R) blocker, on the mitochondria-specific genes expression in a mouse model of Parkinsonism.
Materials and methods: Mice were divided into 5 groups with 6 in each; Group I received 0.5% CMC (control) + saline, Group II received 0.5% CMC + 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (positive control), Group III & IV received MPTP + TEL 3 and 10 mg/kg, p.o. respectively, Group V received TEL 10 mg/kg, p.o. (drug control). MPTP was given 80 mg/kg intraperitoneal in two divided doses (40 mg/kg
Mitochondrial dysfunction plays a crucial role in the Parkinson’s disease (PD)
pathogenesis [1]. Neurons are particularly vulnerable to mitochondrial
dysfunction as they are highly dependent on Electron transport chain (ETC) for energy requirements [2]. The
first event in mitochondrial impairment in PD is the deficiency of Complex I in
the ETC [3, 4]. Dopaminergic SNpc neurons are
susceptible to oxidative stress as they have poorly myelinated long and thin
axons [5].
Renin angiotensin system (RAS) in brain play a crucial role in PD [12]. Activation of angiotensin II type 1 receptor (AT1R) triggers inflammatory pathways which increase ROS leading to dopaminergic cell death [12, 13]. Telmisartan (TEL), a lipophilic antihypertensive drug exerts its neuroprotective activity by blocking AT1R in the brain. In our earlier investigations, we have reported its neuroprotective activity acute and chronic model of Parkinsonism in mouse models [14, 15]. Blockade of AT1R using losartan was shown to exert anti-inflammatory activity in MPTP intoxicated mice model of PD [16, 17] and antioxidant properties against 6-OHDA model [18]. Recently, AT1R and AT2R have been discovered in brain mitochondrial outer and inner membrane, respectively [19]. Alleviation of mitochondrial function was shown to decrease oxidative stress and reduced the incidences of apoptosis in various neurological disorders [20, 21, 22, 23].
Recent understandings of the existence of RAS components in mitochondrial structure, we proposed investigate whether AT1R antagonism modulates mitochondrial functions, particularly in PD. As a preliminary approach, the present experiment is designed to unravel the role of AT1R antagonism using TEL on the mitochondria-specific genes expression in a MPTP model of Parkinsonism in mice.
Male C57BL/6J mouse (18–22 g b. wt.) were obtained from CPCSEA approved breeder
and acclimatized for 7 days before the treatment, with an ambient temperature of
22
Telmisartan API was received as a kind gift from Bal Pharma Limited, Bangalore, India. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and TRIzol reagent were purchased from Sigma Aldrich, USA. PCR master cycler gradient was procured from Genet Bio, Korea. All the chemicals used in the experiment were analytical grade.
Animals were grouped into 6 per group, where Group I (Control) received 0.5%
CMC + Saline as a vehicle, Group II (Positive Control) received MPTP + Saline,
and Group III (TEL LD) & Group IV (TEL HD) received TEL 3 and 10 mg/kg p.o.
respectively + MPTP and Group V received TEL 10 mg/kg p.o. as drug control.
Neurotoxin, MPTP was given 80 mg/kg intraperitoneal in two divided doses (40
mg/kg
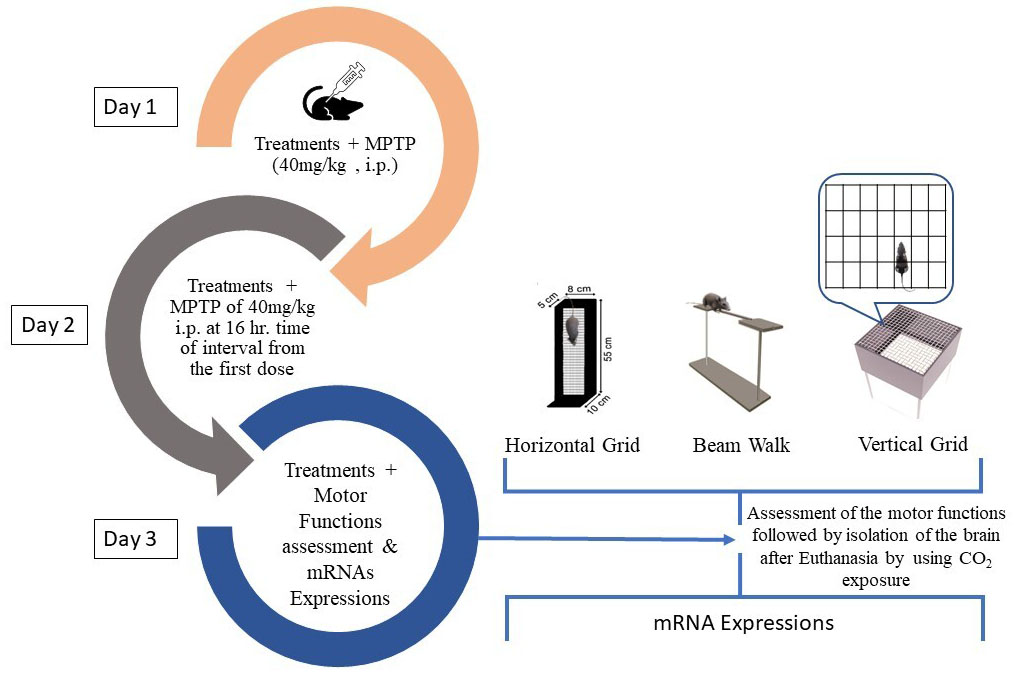 Fig. 1.
Fig. 1.Experimental design for the acute MPTP model of PD.
Beam walk test was done after the MPTP intoxication, to cross a narrow beam of 100 cm length connecting to an escape box [26]. An aversive stimulus was created with an optimistic light (60 lux) above the beam to trigger the mice to a dark goal box on the other side of the beam. The mice were kept at the beginning of the beam after 48th h of the first exposure of MPTP injection. The time taken to reach the escape box on the other hand (sec), number of foot slip (s), and immobility period were noted. The observer was blinded to the treatment protocols.
Horizontal grid test was done as per Kim et al. [27] method with minor
changes. The apparatus was made up of horizontal grid mesh (size 12 cm
Vertical grid test was done to assess the motor function as per the method of
Kim et al. [27] with minor modifications. The apparatus is made of an
open box of size 8
Reverse transcriptase-polymerase chain reaction (RT-PCR) was done to determine
the level of mRNA expression of
| Genes | Forward | Reverse |
| α-syn | 5′-GTTCTTCAGAAGCCTAGGGAGC-3′ | 5′-CCACACCCTGCTTGGTTTTC-3′ |
| Parkin | 5′-AAATGCATCTGGAGGGGACG-3′ | 5′-ACCTCTGGCTGCTTCTGAAT-3′ |
| MTA1 | 5′-GTCAGCCCCTGTCATCAACA-3′ | 5′-GACCCCTACTGGGCATCAAG-3′ |
| DJ-1 | 5′-CATGCGTGCTGGGTACACG-3′ | 5′-CCATCTCCTCTGCTCCTTTGG-3′ |
| UCHL1 | 5′-GCAATGCCTGTGGAACGATT-3′ | 5′-TGACCTTCATGTGCACTGGT-3′ |
| PINK1 | 5′-GTTGCAATGCCGCTGTGTAT-3′ | 5′-GCAAAGGGAAAGGTGGGAGT-3′ |
| LRRK2 | 5′-TGCCATGCACAGATATTCAGC-3′ | 5′-TGCAAGCAGCAAAGTACGTG-3′ |
| GAPDH | 5′-TTCACCACCATGGAGAAGGC-3′ | 5′-TCATGACCACAGTCCATGCC -3′ |
Data obtained in the experiment were expressed as mean
MPTP intoxicated mice showed an increased immobility period and took a long time
in crossing the narrow runway with more numbers of foot slips (P
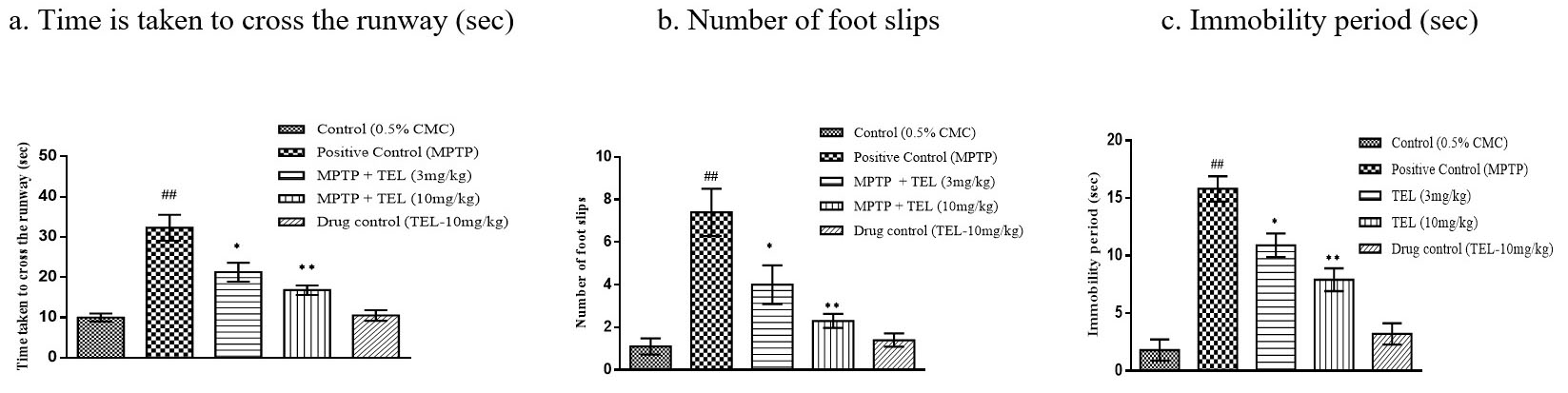 Fig. 2.
Fig. 2.Effect of TEL on time taken to cross the runway (sec), number of
foot slip (s), and immobility period (sec) in beam walk test. * & ** indicates
P
MPTP intoxicated mice showed a longer wall hanging time (P
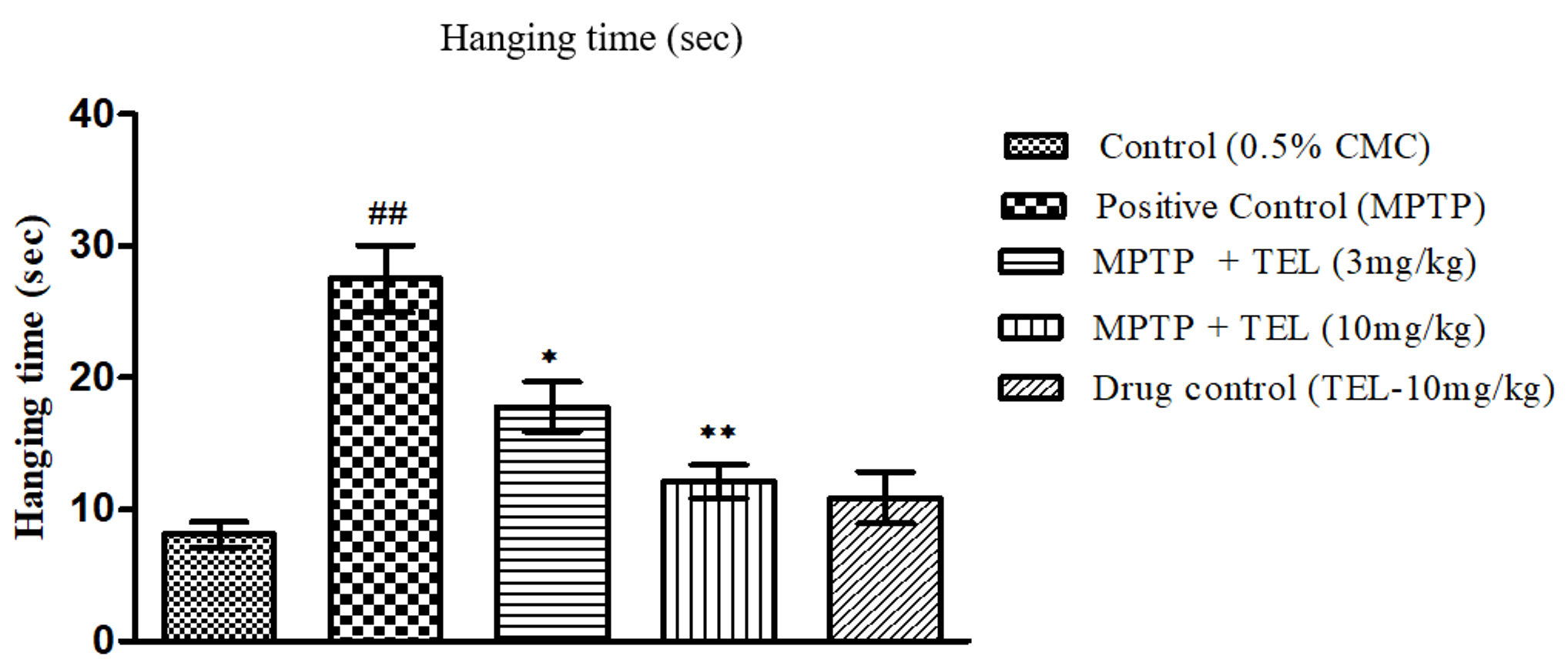 Fig. 3.
Fig. 3.Effect of TEL on the hanging times (sec) assessed on Vertical
grid test. * & ** indicates P
MPTP intoxicated mice took a longer time (P
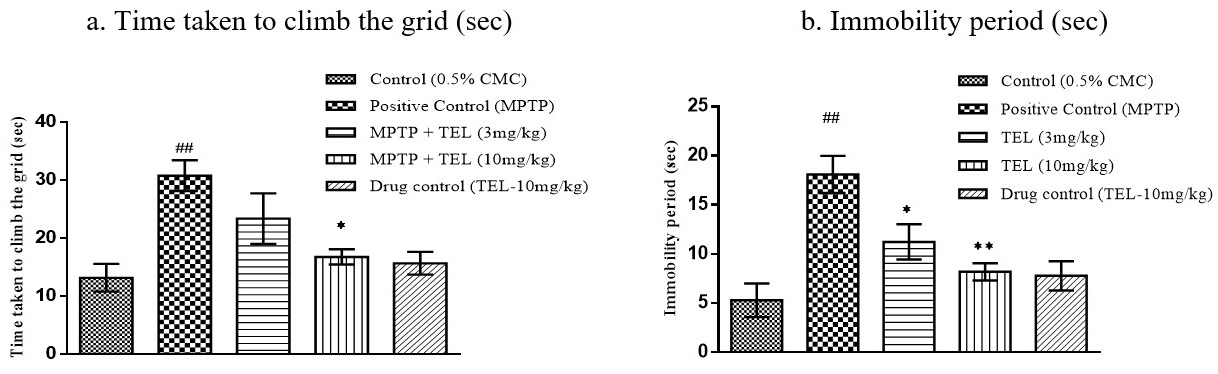 Fig. 4.
Fig. 4.Effect of TEL on time taken to climb the grid (sec), immobility
period (sec) on horizontal grid test. * & ** indicates P
Significant upregulations of
Significant upregulations of
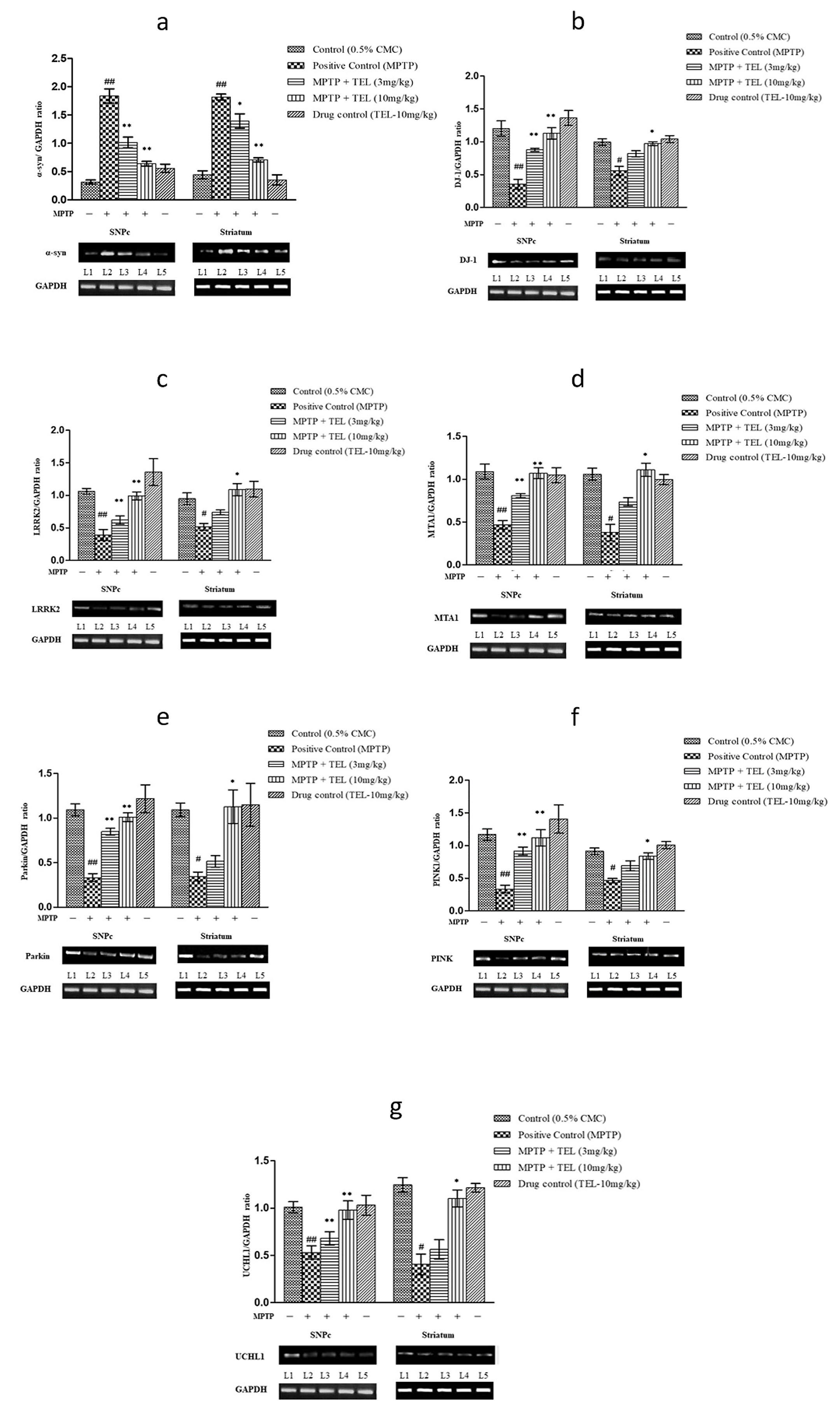 Fig. 5.
Fig. 5.Effect of TEL on gene expressions such (a)
The current study adds further evidence on the neuroprotective role of AT1R antagonism and also new information on its ameliorative effects on mitochondria-specific genes expression in PD. Inhibition of Complex I of ETC was reported in PD patient’s brains [28, 29]. Neurotoxin MPTP produces PD-like conditions by inhibiting the Complex I. Recently, AT1R and AT2R (RAS) were identified on the mitochondrial surface and inner membrane, respectively [19, 30]. Activation of AT1R was shown to impair the mitochondrial functions via NAD(P)H mediated oxidative stress in the PD [31]. In our early study, we have shown that TEL suppresses nitrosative stress and elicits neuroprotection in a mouse model of PD [32]. However, to date, there is no evidence on the effects of AT1R antagonism on mitochondria-specific genes expression which plays a crucial role in oxidative stress, inflammatory responses, and apoptosis. Hence, in the present study we investigated the effects of TEL on the mitochondria-specific genes like PINK1, Parkin, LRRK2, DJ-1 which may serve as newer therapeutic target in the treatment of PD.
Knockout/mutation in PINK1 and Parkin proteins are linked with PD pathogenesis [33, 34, 35]. Parkin is selectively recruited by PINK1 to the outer membrane of the damaged mitochondria to promote mitophagy in PD [36]. In the present study, pre-treatment with TEL upregulated the PINK1 and Parkin in MPTP intoxicated mice brains, which indicates that AT1R antagonism may support the PINK1-Parkin mediated mitophagy and in turn improving mitochondrial dynamics [37, 38].
Knockout/mutation (G2019S) in LRRK2 impair autophagy and aggregation of
In physiological condition,
Finally, the upregulation of MTA1 expression is reported to increase dopamine synthesis [52] and its packaging and subsequent release of dopamine from neuronal vesicles through the upregulation of vesicular monoamine transporter-2 (VMAT2) [53, 54]. The improved motor function recorded in beam walk, horizontal grid, and vertical grid tests with TEL might be due to the increased dopamine levels which may be corroborated by increased expression of MTA1 and DJ-1 in PD mice. The current findings support that modulation of brain RAS function improves mitochondrial function and can impart neuroprotection in PD (Fig. 6).
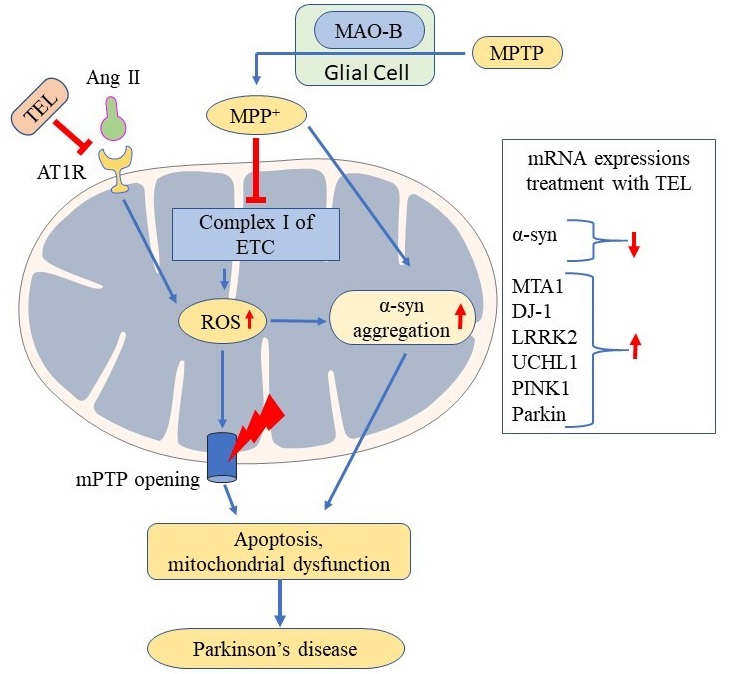 Fig. 6.
Fig. 6.Possible neuroprotective effect of TEL improving mitochondrial functions via inhibiting mitochondrial AT1R in PD.
In conclusion, modulation of central RAS, particularly AT1R antagonism improves mitochondrial functions and exerts neuroprotection in PD.
BR performed the study. GR, SV and SR analyzed the data. ST and AMM assisted the study. MME and SBC designed the study corrected and finalized the manuscript.
Institutional animal ethical committee, Central animal house, JSS Academy of higher education and Research, Mysuru, Karnataka, India approved the study (JSSAHER/CPT/IAEC/016/2020).
BR acknowledges the Indian Council of Medical Research (ICMR), New Delhi, Govt. of India, for the Senior Research Fellowship.
This research received no external funding.
The authors declare no conflict of interest.