
Academic Editors: Massimo Conese and Lorenzo Guerra
Background: Recently, we provided evidence that a single nucleotide polymorphism (SNP), rs41266431, on the gap junction protein alpha 4 (GJA4) gene, acts as a modifier for clinical disease severity in patients with cystic fibrosis (CF). These features are very similar to those of variants of the mannose-binding lectin (MBL). This study aimed to clarify whether the clinical disease phenotype associated with GJA4 variants is independent of MBL variants. Methods: One hundred and twelve patients with homozygous F508del (mean age, 27.6 years; m/f, 61/51) were recruited from the CF centers of Bonn, Frankfurt, and Amsterdam. A sequence analysis was performed for GJA4 and MBL. The clinical phenotype was assessed over three years using pulmonary function tests, body mass index, Pseudomonas aeruginosa colonization, diabetes mellitus, survival to end-stage lung disease, and inflammatory markers. Results: A clinically relevant SNP of GJA4 was identified by sequence analysis. Pulmonary function (FVC% pred, mean 78/85; p
There are various lung disease phenotypes for the delta F508 (homozygous) genotype in cystic fibrosis [1, 2]. Progressive pulmonary destruction is a major cause of morbidity and mortality in patients with CF [3]. Leukocyte-driven inflammation is the most important factor in the pathogenesis of CF lung disease. Previously, we provided evidence that blood cytokines correlate negatively with pulmonary function in F508del-homozygous patients [4, 5, 6].
Mannose-binding lectin is an innate immune protein produced in the liver. It may activate the lectin pathway of the complement system or directly opsonize organisms (e.g., bacteria, viruses, and fungi) to enhance phagocytosis. Although MBL is primarily a serum protein, it accumulates in the lungs during acute inflammation [7]. Recent evidence suggests an important role of MBL in patients with CF [7].
MBL2 genotypes are associated with CF lung disease severity, including the earlier acquisition of Pseudomonas aeruginosa (p
Recently, we provided evidence that one single nucleotide polymorphism, rs41266431, on the gap junction protein alpha 4 gene causes an amino acid substitution with a clinical impact, making carriers of the A allele have significantly better protection against end-stage lung disease and better pulmonary function. This genotype has no impact on the frequency of P. aeruginosa colonization, but those who are carriers of the A allele and chronically colonized with P. aeruginosa have significantly better pulmonary function than those with the G/G genotype [10].
Regarding this matter, whether there is a link between the MBL variant alleles and the GJA4 variants should be elucidated as the clinical disease phenotypes are similar. Moreover, to the best of our knowledge, there are no data on MBL variant alleles in Dutch patients and only in a small sample of German patients [11]. Accordingly, this study aimed to clarify whether the clinical disease phenotype associated with GJA4 variants is independent of MBL variants.
For this exploratory study, delta F508 homozygous patients were recruited from the CF center of the Children’s Hospital Medical Center at the University of Bonn. For replication studies, patients were recruited from the CF centers of Goethe-University Frankfurt and the Department of Respiratory Medicine, Amsterdam University Medical Centers (Amsterdam UMC).
Before inclusion in the study, a detailed verbal and written explanation was provided to all the patients and their caregivers. The course of the study, as well as its goals and risks, were discussed in detail. Prior to study initiation, the participants or their legal guardians signed a consent form. The protocol was approved by the ethics committees of the University of Bonn (178/01 + 092/17), Frankfurt (07/02 + 206/16), and Amsterdam UMC (NL60220.018.16). Informed consent was obtained from all patients or their parents. The study was registered with ClinicalTrials.gov (NCT04242420) retrospectively on 24 January 2020 and updated on 24 November 2020.
The exclusion criteria were treatment with systemic steroids 14 days preceding this trial, participation in another study within the past 30 days, treatment with a lumacaftor/ivacaftor combination (Orkambi), poor status after lung transplantation, and inability to perform all study procedures.
All patients underwent spirometry (forced expiratory volume in one second [FEV1], forced vital capacity [FVC], and forced expiratory flow at 75% of the pulmonary volume [FEF75]) and supplied a blood sample (EDTA and serum). Pulmonary function tests were performed according to the recommendations of the American Thoracic Society and European Respiratory Society [12].
Serum MBL levels were determined using an enzyme-linked immunosorbent assay (Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA). According to the manufacturer, this assay has a high intraassay precision (CV
According to Garred [9], in exon 1 of the MBL2 gene, three single-base substitutions independently cause low serum levels of MBL at codon 54 (glycine with aspartic acid, allele B), codon 57 (glycine with glutamic acid, allele C), and codon 52 (arginine with cysteine, allele D) [13, 14, 15]). The common designation for these variant alleles is 0, whereas the normal allele is A. Several nucleotide substitutions in the promoter region of the MBL2 gene also affect the MBL serum level. In particular, a polymorphism in codon 221 (X/Y type) has a significant downregulating effect on MBL serum concentration [16, 17, 18]. We pooled the genotypes with low serum MBL levels (0/0, XA/0, YA/0) according to Garred [16] into the MBL-insufficient group and those with high serum MBL levels (XA/XA, YA/XA, YA/YA) into the MBL-sufficient group.
For gene analysis, standard procedures were used for the isolation of genomic DNA, amplification of DNA via polymerase chain reaction (PCR), and automated sequencing analyses. Briefly, oligonucleotides (forward primer MBL2-PF: TATTTAGCACTCTGCCAGGGC and reverse primer MBL2-1R: CAGTCTCCTCATATCCCCAGG) were used to amplify 950 bp fragment-covering parts of the MBL gene promoter and its exon 1. This PCR product harbors all relevant variants reported by Madsen [17] and Garred [9] (Table 1, Ref. [9, 17]).
| (H/L, -550) | (-427) | (F/G, -349) | (J/K, -336) | (M/N, -329 to 324) | (Y/X, -221) | - | - | (codon 52) | (codon 54) | (codon 57) |
| rs11003125 | rs11003124 | rs7084554 | rs36014597 | rs45560739 | rs7096206 | rs11003123 | rs7095891 | rs5030737 | rs1800450 | rs1800451 |
| c.-619C |
c.-496A |
c.-418A |
c.-405A |
c.-401_-396 delAGAGAA | c.-290 C |
c.-139C |
c.-66C |
c.154C |
c.161G |
c.170G |
| p.Arg52Cys | p.Gly54Asp | p.Gly57Glu | ||||||||
| MBL gene variants [9, 17] and were covered by our 950 bp PCR fragment investigated. The first line gives the initial term for the variant coined by the respective author, with its initial nucleotide position; the corresponding rs number is given below, and line three gives the actual nucleotide position according to UCSC genome browser GRCh38/hg38. | ||||||||||
In this study, the first line gives the initial term for the variant coined by the respective author, with its initial nucleotide position; the corresponding rs number is given below, and line three gives the actual nucleotide position according to the UCSC genome browser GRCh38/hg38.
The resultant PCR products were subjected to direct automated sequencing (3130XL Genetic Analyzer; Applied Biosystems, Foster City, CA, USA). For each patient, both strands of the amplicons were sequenced, and PCR primers were used as sequencing primers. All analyses were performed in the genetic laboratory at Bonn (Institut für Klinische Chemie).
Genotyping for the variant rs41266431 was performed as previously published [10] and in the same cohort. However, in our previous study, the cohort included 116 patients. Because we were only able to explore MBL variants in 112 patients, we analyzed GJA4 variants in these 112 patients. This was done to optimize the equality of the cohort analyzed for GJA4 and MBL pheno/genotype relationships and gene-gene interactions.
Pulmonary function tests, that is, FEV1, FVC, and maximum expiratory flow at 25% of FVC (FEF75), were performed using a Master Screen Body or IOS (Vyaire Medical GmbH, Wuerzburg, Germany) in Bonn and Frankfurt. Carefusion Jaeger® Pneumo Vyntus (Vyaire Medical, Houten, The Netherlands) was used in Amsterdam. As lung volume is dependent on height and age, pulmonary function data were presented as the percentage predicted for height, sex, and age. To accurately assess individual lung function, the median pulmonary function test results over a 3-year period as a predicted percentage of the global lung initiative (GLI) values were acquired for German patients. Data were obtained from the German CF registry (MUKOWEB, www.mukoviszidose-register.de). For the Bonn cohort (Step 1 registry data), only one value per year was available.
For most Frankfurt patients, Step 2 registry data with more than one visit at the CF center per year were available. For these patients, the visit with the best pulmonary function data for FEV1 within a given year was selected. The median age was calculated as the second year of the three observation years. As the Dutch lung function data were cross-sectional (one measurement for 2017 available), and German data were longitudinal (if possible, 2018–2016), the German cohort was analyzed separately.
We compared the aggregated outcomes (median over 3 years) of continuous data between groups. For parametric data, means, standard deviations, and 95% confidence intervals (CIs) were calculated and tested using student’s t-test. Non-parametric data are presented as medians and interquartile range (IQR) and were tested using the Mann-Whitney U-test for unpaired samples. Sputum culture results were used to categorize patients in P. aeruginosa-positive or -negative individuals. The binary data were analyzed using the chi-square test. Survival analysis was performed using a Kaplan-Meier analysis. Moreover, a mixed linear model was used to estimate the effect of MBL and GJA4 genotype and chronic P. aeruginosa colonization on pulmonary function. The additional multivariate analysis included covariates, such as age and body mass index (BMI). For cross-sectional data (such as serum MBL), a general linear model was chosen to estimate the effect of MBL and GJA4 genotype and chronic colonization on serum MBL levels.
A p-value
A total of 112 delta F508 homozygous patients (61 male and 51 female Caucasian patients, (p
Genotyping was performed for all the patients. Of the 112 patients, 79 (71%) were classified as MBL-sufficient and 33 (29%) as MBL-insufficient.
For the GJA4 variant (rs41266431), patients were grouped into those homozygous for the G allele (G/G genotype, n = 82, 73%) and carriers of the A allele (A/G + A/A genotype, n = 30, 27%). Patients were classified as high risk for more severe clinical disease (n = 24, 21%) if they were carriers of the risk diplotype for MBL and GJA4. Patients were considered to have intermediate risk (n = 67, 60%) if they had one protective and one risk variant. If patients were carriers of protective variants of MBL and GJA4, their risk for more severe disease was considered low (n = 21, 19%). Patient characteristics according to haplotypes are presented in Tables 2,3 shows the characteristics of a subset of the matched pairs of adults.
| Genotype | MBL | GJA A4 (rs41266431) | Risk haplotype | ||||
| Sufficient | Insufficient | (G/G) | (A/G; A/A) | High | Intermediate | Low | |
| (n = 79) | (n = 33) | (n = 82) | (n = 30) | (n = 24) | (n = 67) | (n = 21) | |
| Mean | |||||||
| Age (years) | 28.9 |
24.4 |
26.7 | 29.83 | 25.3 |
26.6 |
33.2 |
| SEX (m/f) | (46/33) | (15/18) | (44/38) | (17/13) | (11/13) | (37/30) | (13/8) |
| P.aeruginosa+ (%) | 67% | 73% | 66% | 76% | 71% | 66% | 75% |
| Diabetes mellitus |
33% | 32% | 33% | 32% | 35% | 32% | 36% |
| BMI (percentile) | 35.2 |
23.8 |
29.7 | 37.7 | 19.3% | 34.3% | 38.5% |
| FEV1 (% predicted) | 66 | 68 | 65.5 | 69.4 | 65.6 | 66.9 | 66.6 |
| FEF75 (% predicted) | 53.2 | 61.4 | 54.2 | 59 | 61 | 52.9 | 57.9 |
| FVC (% predicted) | 79.7 | 80.2 | 78 |
85.4 |
77.7 | 79.1 | 84.6 |
| MBL (Serum (ng/mL)) |
820 |
372 |
533 |
365 |
424 | 589 | 762 |
| Interleukin-8 (pg/mL) |
10.6 | 9 | 10.8 | 9.0 | 11.25 | 9 | 10.55 |
| CRP (mg/dL) |
0.185 | 0.365 | 0.24 | 0.23 | 0.34 | 0.2 | 0.175 |
| Genotype | MBL | |
| Sufficient | Insufficient | |
| n = 22 | n = 22 | |
| Mean | ||
| Age (years) | 25 | 25 |
| SEX (m/f) | (11/11) | (11/11) |
| P.aeruginosa+(%) | 68 | 73 |
| Diabetes mellitus |
41 | 32 |
| Bronchiectasis (%) | 77 | 91 |
| BMI (percentile) | 33 |
13 |
| FEV (% predicted) | 69 | 65 |
| FEF75 (% predicted) | 44 | 48 |
| FVC (% predicted) | 82 | 76 |
| m, male; f, female; P. aeruginosa +, Pseudomonas aeruginosa colonization (intermittent+chronic); FEV1 (% predicted), forced expiratory volume in 1 s in the predicted percentage; FEF75 (% predicted), forced expiratory flow at 75% of the pulmonary volume; FVC (% predicted), forced vital capacity in the predicted percentage; | ||
There was no difference in the observation time for the two genotypes (MBL-sufficient/insufficient: mean 31.5/28 years; 95% CI, 28.4–34.5/24–31.2 [range, 9–62/9–54] years; p
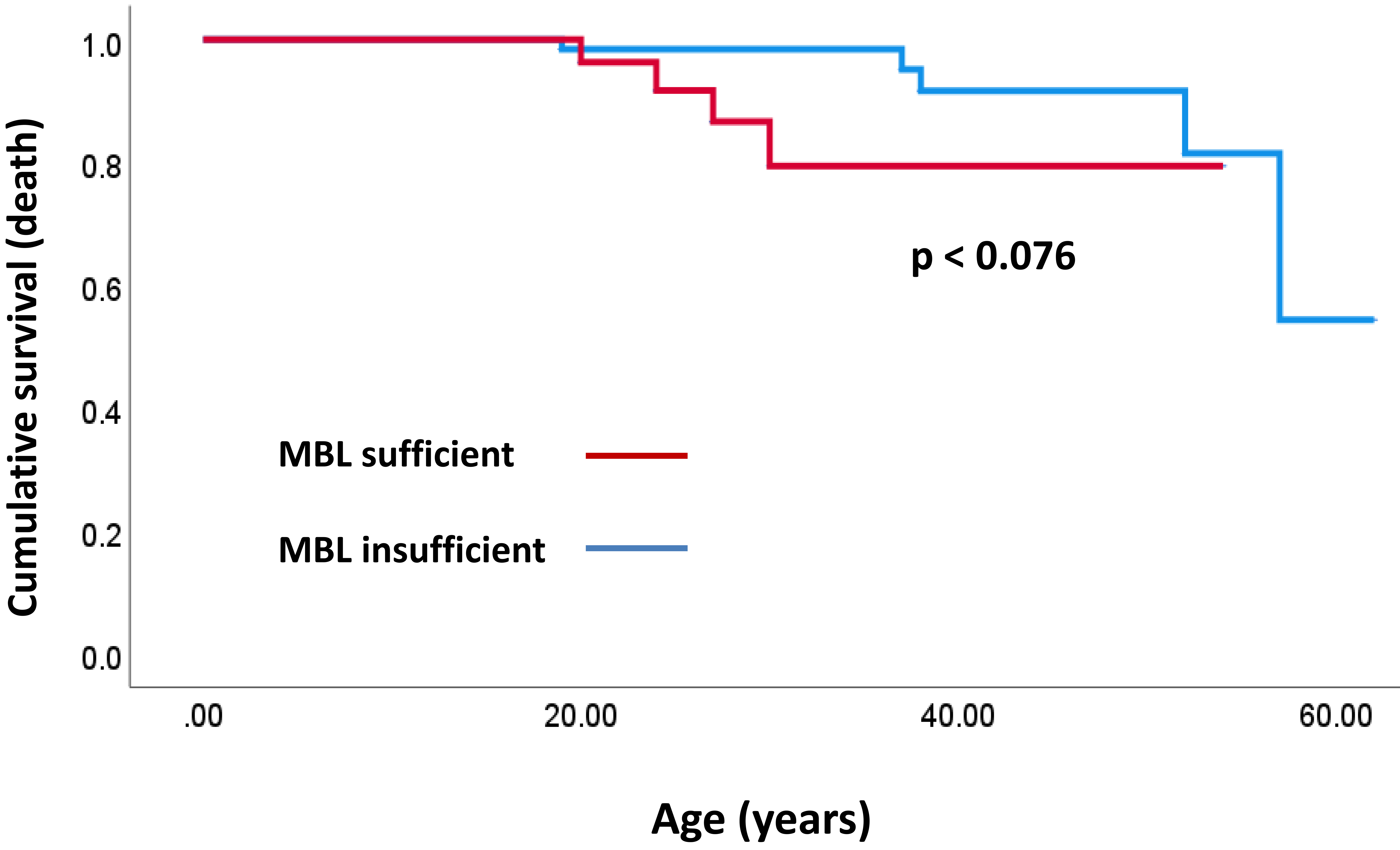 Fig. 1.
Fig. 1.Kaplan-Meier plot of the survival of patients with CF. An event was defined as the age at death (n = 9). The red line indicates MBL-sufficient variants and the blue line indicates MBL-insufficient variants.
The Cox regression analysis revealed a 3.4-fold (CI 0.814–13.9) higher risk of mortality for those with the MBL-insufficient genotype compared to that of the overall cohort (p
Overall, up to 2018, 13 patients who did (n = 4) or did not (n = 9) receive a lung transplant died, meaning that four MBL-insufficient patients (12%) and nine MBL-sufficient patients (11%) (p
End-stage lung disease: Ten MBL-sufficient (13%) and five MBL-insufficient (15%) patients were alive after lung transplantation. The log-rank test did not reveal a significant difference in survival (p
There was no difference in observation time (G/G genotype/ [A/G; A/A genotype]; mean, 29.4/32.9 years; 95% CI, 26.7–32.1/27.8–38.1 [range, 9–58/13–62 years]; p
Overall, until 2018, in addition to nine deaths among patients with CF without lung transplantation, four patients underwent lung transplantation, resulting in 13 deaths among G/G genotype carriers and no deaths among carriers of the A allele (p
Moreover, in the entire cohort, six lung transplants were performed, with two patients still alive. In this regard, 15 patients were classified as having end-stage CF, that is, death or lung transplant. Fourteen patients had the G/G genotype, and one patient with CF alive after lung transplantation was a carrier of the A allele. Thus, end-stage CF was significantly more common in the G/G genotype group (p
A Cox regression analysis revealed a seven-fold (CI, 0.917–54.136) higher risk for those with the G/G genotype to experience severe lung disease than the overall cohort (p
There was no difference in the observation time between the three haplotypes (low/intermediate/high: mean, 35.5/29.6/27.9 years; 95% CI, 28.6–42.5/26.6–32.5/23.1–32.7 [range, 13–62/9–58/9–54] years; p
A Cox regression analysis revealed a 4.3-fold (CI, 1.3–13.8) higher risk of mortality for those with intermediate or high risk compared to that of low-risk patients (p
End-stage lung disease: One patient at low risk (5%), nine at intermediate risk (13.4%), and five (21%) at high risk were alive after lung transplantation. End-stage CF was more common in high-risk patients (p
Seventy-six (68.5%) patients showed microbiological evidence of P. aeruginosa colonization (chronic, n = 57 [51.4%]; intermittent, n = 19 [17%]).
The MBL genotype (MBL-sufficient vs. -insufficient) did not have an impact on colonization (intermittent + chronic): MBL-sufficient, n = 52 (67%) vs. MBL-insufficient, n = 24 (73%) (p
To determine the impact of P. aeruginosa on clinical course, longitudinal data, which were only available from the German cohort, were evaluated. The characteristics of the patients with chronic P. aeruginosa colonization are shown in Table 4.
| Genotype | MBL | GJ4 A4 (rs41266431) | Risk haplotype | ||||
| Sufficient | Insufficient | (G/G) | (A/G;A/A) | High | Intermediate | Low | |
| n = 33 | n = 15 | n = 35 | n = 13 | n = 11 | n = 27 | n = 10 | |
| Mean | Mean | Median | |||||
| Age (years) | 37 |
29 |
34 | 38 | 28 |
34 |
48 |
| SEX (m/f) | (23/10) | (10/5) | 23/12 | 10/3 | 7/4 | 18/9 | 8/2 |
| Diabetes mellitus | 49% | 47% | 51% | 39% | 55% | 46% | 44% |
| BMI (percentile) | 21 |
10 |
18 | 15 | 3 | 16.5 | 4 |
| FEV (% predicted) | 52 | 48 | 48 | 59 | 37 | 46 | 62 |
| FEF75 (% predicted) | 28 | 26 | 26 | 30 | 19 | 20 | 32 |
| FVC (% predicted) | 71 | 62 | 65 |
78 |
56 |
69 |
84 |
| MBL (Serum (ng/mL)) | 556 |
456 |
492 | 556 | 474 | 474 | 968 |
| IL-8 (pg/mL) |
9 | 10.6 | 10.6 | 7 | 16.3 | 8.3 | 8.5 |
| CRP (mg/dL) |
0.3 | 0.47 | 0.42 | 0.23 | 0.43 | 0.425 | 0.175 |
The GJA4 genotype did not have an impact on colonization ever (intermittent + chronic): G/G genotype, n = 54 (65.9%) vs. carriers of the A allele, n = 22 (75.9%) (p
The risk haplotypes (high vs. intermediate vs. low) did not have an impact (p
To determine the impact of P. aeruginosa on the clinical course, longitudinal data, which were only available from the German cohort, were evaluated. The characteristics of the patients with chronic P. aeruginosa colonization are shown in Table 4.
There was no evidence on the influence of MBL or the gap junction protein alpha 4 genotype on the age at first acquisition and mucoid conversion of P. aeruginosa.
The spirometric data on FEV1, of all 112 patients were available, for 107 on FVC, and FEF75 (Table 2). In the adult subcohort, there was a significant difference in age (p
BMI percentiles were available for all 112 patients. In the overall cohort, there was a trend for a difference (p
Serum MBL levels were significantly higher for the MBL-sufficient genotype (MBL-sufficient/-insufficient genotype: median, 820/372 mg/mL; interquartile range [IQR], 353–1310/254–474; p
The results of the general linear model, which was adjusted for age and chronic colonization with P. aeruginosa, indicated a higher serum MBL (estimated, +503 ng/mL; 95% CI, 188–818; p
The findings of this study provide evidence that the clinical disease phenotype associated with GJA4 variants is independent of MBL variants. A novel finding of this study is that patients with CF with MBL-sufficient genotypes had significantly better BMI percentiles than those with insufficient MBL. This is a very interesting finding since patients with CF have an increased risk of sarcopenia and osteopenia compared to the general population. However, a well-balanced nutritional status may reduce the risk of respiratory and metabolic complications.
We created risk haplotypes to investigate the gene-gene interactions between MBL and GJA4 variants. The GJA4 risk genotype was linked to significantly worse lung function (FVC) in the overall cohort. However, the evaluation of risk haplotypes for gene-gene interactions did not reveal any augmentation of these effects. For example, the high-risk haplotype did not have significantly worse lung function than the other risk variants.
Though other studies [9, 19] were able to show the impact of MBL variants on lung function, we were unable to find it in this cohort of German and Dutch patients with CF. To the best of our knowledge, there is only one other small study (n = 35) on delta F508 homozygous carriers in Germany. They found that MBL insufficiency leads to a shorter interval between the first PA infection and onset of chronic infection. However, irrespective of the PA infection status, the FEV1% values did not vary significantly between MBL-sufficient and MBL-insufficient producers in their study [11].
Moreover, studies on the effects of MBL variants on the lung disease phenotype showed conflicting results in similar ethnicities in Northern America (Canada and USA). Dorfman [20] found an effect on lung function, but two other studies did not [21, 22]. A meta-analysis by Chalmers [7] revealed that a negative effect on lung function is rather shown in adult cohorts but not in pediatric cohorts. Even with the exclusion of children (age
McDougal and coworkers [22] evaluated lung function data from twin and sibling studies. They did not find any influence of MBL variants. However, they found an association between the first infection of P. aeruginosa and the time to conversion to a mucoid phenotype. However, we did not observe such an association. A meta-analysis of the impact of MBL genotypes associated with MBL insufficiency in CF patients [7] revealed not only an earlier acquisition of Pseudomonas aeruginosa and reduced pulmonary function, but also an increased rate of death or requirement for lung transplantation.
In this regard, our data are consistent with the findings of the meta-analysis. There was a trend for more cases of deaths in the MBL-insufficient group (12%) than in the sufficient-group (6%) in the Kaplan-Meier (p
One of the limitations of this study is the small sample size (n = 112) compared to the twin and sibling study (n = 788) by McDougal [22]. Nevertheless, some studies investigated the same gene variants with similar and even lower number of patients, confirming the findings of the meta-analysis of Chalmers [7] regarding lower lung function. Garred investigated 149 patients with CF in Denmark [9], and Gabolde investigated 22 patients in France [19]. In addition to the potential impact of ethnicity, environmental influences in different countries, including climate and health care practice, and differences in CFTR genotypes and age may account for the different results in the studies mentioned.
Interestingly, there was a significant difference in the BMI percentile between the MBL-sufficient and MBL-insufficient genotypes. To our knowledge, this has not been previously reported in patients with CF. Lower MBL levels could account for an upregulated inflammatory state in CF. In our study, the median CRP levels were higher for the low producer genotype, though this difference was insignificant. It is known that CRP as a BMI is linked to survival in CF [23, 24]. In this regard, we can speculate as to why we found a trend for superior survival due to higher BMI percentiles in MBL-sufficient patients with CF, even without better lung function parameters. Our set of inflammatory data in serum MBL/interleukin-8/CRP was small (n = 52/67/44) and cross-sectional. We cannot exclude the possibility that a wider array of inflammatory parameters, larger samples, and longitudinal data could identify the functional link between BMI and MBL variants.
McDougal [22] demonstrated that YO/YO and XA/YO individuals can be considered to have a MBL “deficiency”, whereas YA/YO, XA/XA, XA/YA, and YA/YA individuals have a high enough level of MBL to be considered “sufficient”. Garred considered the YA/YO variant, which had very low serum MBL levels in their study, as MBL-sufficient, suggesting that this variant had similar lung function to those with high serum MBL levels. As lung function was not a discriminating measure in our cohort, we considered YA/YO variants with low-serum MBL levels to be MBL-insufficient. To compare our results with other studies, such as that of McDougal, we analyzed our data with their haplotyping scheme as well. We classified YA/YO individuals as MBL-sufficient) (Supplementary Table 1). The disadvantage was that we observed a significant difference (p
In summary, our data showed that the clinical disease phenotype associated with GJA4 variants is independent of MBL variants. MBL-sufficient variants were associated with superior BMI and a trend toward decreased mortality in this subcohort.
JPL—investigation, data curation, and writing–original draft and review. ML—conceptualization, methodology, investigation, resources, writing-original draft (lab part) and review, and supervision (genetic lab). TH—investigation. OE—investigation (inflammatory markers) and writing, review, and editing. CS—investigation and writing, review, and editing. RS—investigation, methodology (inflammatory markers), supervision (lab), resources, and writing–review and editing. SZ—conceptualization (inflammatory markers), writing, reviewing, and editing. CM—investigation, writing, review, and editing. MA—writing, review, and editing. SS-G—conceptualization, methodology, data curation, formal analysis, funding acquisition, writing-original draft and review, supervision, and project administration. All authors have contributed to the manuscript and approved the submitted version.
Studies involving human participants were reviewed and approved by the University of Bonn (178/01 + 092/17), University of Frankfurt (07/02 + 206/16), and Amsterdam University Medical Center (NL60220.018.16). Written informed consent to participate in this study was provided by the participants’ legal guardian or next of kin. Clinical Trial Registration: The study was registered with ClinicalTrials.gov (number NCT04242420) retrospectively on January 24, 2020 and updated on November 24, 2020.
The authors acknowledge support from the following: Pia Uerdingen (for technical assistance), Institut für Klinische Chemie und Klinische Pharmakologie des Universitätsklinikums Bonn, Germany. Doris NGampolo (for technical assistance), University Children’s Hospital Bonn, Germany. R. Lub, L. van der Schaaf, M. van Brederode, Y.W.F Dagelet, Dr. E.J.M. Weersink, Department of Respiratory Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands. T. Dekker and B. S. Dierdorp, Department of Experimental Immunology (Amsterdam Infectin & Immunity Institute), Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands.
This work was funded by the heritage of Juliana Gerner, Germany.
(1) Sabina Schmitt-Grohé is on the advisory committee of Vertex and has received a travel grant from Vertex (for ECFS 2019) and ERS (ERS Research Seminars). Moreover, she gave a talk to Vertex, participated in studies for the same, received a fee for a congress report (ECFS conference Liverpool 2019), and attended a vertex-sponsored conference (1.CF-Akademie, Schloss Hohenkammer). In addition, she got financial reimbursement for a talk for Deutsche CF Hilfe and received a personal fee for an expert opinion for the Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen.
(2) Zielen reports grants and personal fees from Bene-Arzneimittel GmbH, grants and personal fees from Biotest GmbH, grants from Vifor Pharma Deutschland GmbHÐ, grants from ALK Arzneimittel, personal fees from Novartis GmbH, personal fees from Böhringer Ingelheim, personal fees from Lofarma GmbH, personal fees from IMS HEALTH GmbH and Co., personal fees from GSK, Stallergen, Procter and Gamble, and Allergopharma GmbH, grants and Allergy Therapeutics, Engelhard Arzneimittel, Sanofi-Pasteur, AstraZeneca, Erydel, and Bionorica GmbH, outside the submitted work.
The other authors declare no conflict of interest.