
- Academic Editor
Background: Apoptosis and pyroptosis are two types of programmed cell death related to the neuroinflammatory reaction after subarachnoid hemorrhage (SAH). Research indicates that triggering receptor expressed on myeloid cells 2 (TREM2) can regulate the SAH-induced inflammatory response. However, whether TREM2 regulates programmed cell death (apoptosis and pyroptosis) remains to be clarified. The purpose of the present study was to investigate the effects of TREM2 on cell death in SAH. Methods: SAH was induced in adult male C57BL/6J mice by endovascular perforation. An in-vitro cellular model of SAH was established by treating cocultured BV2 microglia and HT22 neuronal cells with oxyhemoglobin. TREM2 overexpression or knockdown was carried out by intraventricular lentivirus injection at 7 d before SAH induction in mice or lentiviral transfection, respectively. Neurobehavioral tests as well as western blot, reverse transcription–quantitative polymerase chain reaction (RT-qPCR), immunofluorescence, Evans blue (EB) staining, Nissl staining, and flow cytometry assays were performed to investigate the neuroprotective role of TREM2 after SAH. Results: After SAH, the TREM2 mRNA and protein levels were elevated in SAH mice, exhibiting a peak at 72 h. TREM2 overexpression improved the SAH-induced neurological deficits in mice, while TREM2 knockdown worsened them. In the brains of mice with TREM2 overexpression, less neuronal death and more neuronal survival were detected at 72 h post SAH. Meanwhile, TREM2 overexpression showed an inhibitory effect on microglial activation, neutrophil infiltration, and the expression of cell death marker proteins. Consistent results were obtained in vitro. Conclusions: Our research indicates the important role of TREM2 on cell death after SAH, suggesting that targeting TREM2 might be an effective approach for treating SAH.
Subarachnoid hemorrhage (SAH), which is bleeding in the subarachnoid space, is a common cerebrovascular disease. The symptoms of SAH include headache, vomiting, unconsciousness, numbness, and even seizures [1]. The overall crude incidence of SAH globally is reported to be 6.2–10.0 per 100,000 persons [2]. Up to 30% of SAH patients have a poor outcome or even death, while most survivors suffer from long-term disability or cognitive impairment [3]. SAH may result from a traumatic brain injury or a spontaneous aneurysm rupture. Family history, smoking, alcoholism, and high blood pressure have been confirmed to be the main risk factors for spontaneous SAH [4]. SAH patients are often diagnosed by computed topography and managed by stabilization and prevention of rebleeding, mostly symptomatic treatment. Many mechanisms have been proposed to explain the brain injury damage following SAH. For cerebral vasospasm, which is one typical complication of SAH, blood products are thought to be released from the SAH, which trigger the activation of the tyrosine kinase pathway and cause calcium ion release, making the smooth muscle of the cerebral arteries contract [5]. Besides, oxyhemoglobin released into the cerebrospinal fluild (CSF) can also increase the release of free radicals, endothelin-1, prostaglandin, etc., resulting in vasoconstriction. In addition, studies have shown that the inflammatory reaction, which is featured by microglial activation, inflammatory cell infiltration, and cytokine release, contributes to the pathogenesis of SAH-induced brain injury [6]. Furthermore, oxidative stress, mitochondrial dysfunction, etc. also have been widely accepted as detrimental factors in SAH-induced brain injury [7]. However, based on the current situation that SAH patients still have poor neurological function outcomes, more investigations are needed to clarify the detailed mechanism.
The triggering receptor expressed on myeloid cells 2 (TREM2), which is
selectively and highly expressed on microglia, is the main receptor for inducing
the anti-inflammatory response [8]. It can be activated by damage-associated
molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs),
and then it can combine with adaptor proteins, triggering the immune responses in
microglia [9, 10]. TREM2 also can be regulated by nuclear factors, which play an
important role in inflammation [11]. Many researchers have reported the
neuroprotective effect of TREM2 by promoting phagocytosis and suspending
inflammation in experimental intracerebral hemorrhage [12, 13]. Moreover, TREM2
overexpression can decrease the expression of proinflammatory cytokines, such as
interleukin (IL)-1
Apoptosis and pyroptosis are two types of programmed cell death related to the
neuroinflammatory reaction in intracranial hemorrhagic disease [21, 22]. The
activation mechanisms of apoptosis include both intrinsic and extrinsic pathways.
Generally, when apoptosis begins, the TNF-
The present study aimed to clarify the effects of TREM2 in neuroinflammation and programmed cell death in in-vivo and in-vitro SAH models. By using lentiviral transfection for bidirectional intervention of TREM2 expression, we compared the results of TREM2 knockdown and overexpression on the cognitive status, neuroinflammatory response, and programmed cell death in mice. We hypothesized that TREM2 activation would attenuate the neuroinflammatory reaction and interfere with programmed cell death as well as alleviate brain injury in experimental SAH; therefore, TREM2 may serve as a pharmacological target in SAH therapy.
Male C57BL/6J mice (6–8 weeks old, 20–25 g) were from GemPharmatech Co., Ltd. (Nanjing, China). All mice were housed under a 12-h light/dark cycle and had access to food and water ad libitum. Animal handling and all of the related experimental procedures were carried out according to the National Institutes of Health guidelines and approved by the Animal Ethics Review Committee of Wannan Medical College (approval number: WNMC-AWE-2023293).
To explore the expression pattern and localization of TREM2 in the brain, mice were randomly divided into six groups: Sham, SAH 1 d, SAH 2 d, SAH 3 d, SAH 5 d, and SAH 7 d (n = 6). To evaluate the neurological function in mice after SAH, mice were randomly divided into four groups: Sham, SAH, SAH+sh-NC, and SAH+sh-TREM2. To verify the role of TREM2 in programmed cell death, mice were randomly divided into four groups: Sham, SAH, SAH+sh-NC, and SAH+sh-TREM2. The brain water content, open field test, neurological evaluation, western blot, reverse transcription–quantitative polymerase chain reaction (RT-qPCR), and immunofluorescence assays were performed at 72 h after SAH.
The SAH model was built via endovascular perforation, as reported previously
[28]. The animals were first anesthetized by isoflurane (Sigma-Aldrich, St.
Louis, MO, USA) inhalation. Then, the right common carotid artery, internal
carotid artery, and external carotid artery were exposed clearly under a
microscope. A MACO nylon suture (0.2
At 72 h after the operation, the animals were evaluated by the modified Garcia Neuroscore [29, 30]. The Garcia Neuroscore includes six subtests: spontaneous activity, limb extension, forepaw outstretching, climbing, side stroking, and vibrissae touch. The mice were graded, and the total score was calculated, ranging between 0 (greatest deficits) and 18 (without deficits).
The open field test was performed in a 40 cm
The mice were injected with 0.4 mL of 1% EB (Acmec Biochemical Technology (ACMEC), Shanghai, China) solution through the tail vein and sacrificed 0.5 h later. A 100-mg sample of brain tissue was taken and homogenized. After centrifugation at 1000 g for 15 min, the supernatant was mixed with acetone (supernatant:acetone = 3:7) and incubated at room temperature for 24 h. The centrifuged supernatant (2000 g, 15 min) was used for the determination of the optical density at 620 nm. The content of EB was calculated according to the EB standard curve.
The mice were sacrificed at 72 h after SAH surgery, and the brains were removed.
The wet weight (W) was immediately recorded. Then, the specimens were put in an
oven at 105 °C for 24 h, and the dry weight (D) was also recorded. The
brain water content was calculated by the following formula:
[(W–D)/W]
The mouse brain tissues were put in 4% paraformaldehyde for 72 h after dissection. Then, the tissues were embedded with paraffin and cut into sections (4 µm). Nissl staining (Beyotime, Shanghai, China) was performed according to the manufacturer’s instructions. Normal neurons have relatively big cell bodies and are rich in cytoplasm, with one or two big round nuclei, whereas damaged cells have shrunken cell bodies, condensed nuclei, a dark cytoplasm, and numerous empty vesicles.
Microglial BV-2 cells were cultured in Dulbecco’s modified Eagle medium.
Lentivirus (Hanbio Co., Ltd. Shanghai, China) bearing TREM2 shRNA or the
whole-length TREM DNA sequence was used for the construction of cell lines with
TREM2 interference or overexpression, respectively. The sequence of siRNA used
for TREM2 knockdown was ACAGTCATCGCAGATGACACCCTTG. TREM2 overexpression was done
with Mouse-tagged ORF Clone Lentiviral Particle (NM-031254, NCBI). Lentiviral
transfection was screened by puromycin for 2–3 passages until the establishment
of stably transfected cell lines. All cell lines were validated by short tandem
repeat profiling and tested negative for mycoplasma. Cells were all cultured in a
humidified incubator at 37 °C and 5% CO
BV-2 microglial cells and HT22 neuronal cells were cocultured by using polycarbonate membrane transwell chambers (pore size of 1.0 µm, JET BIOFIL, Guangzhou, China). BV2 cells were placed in the upper chamber (interfering cells), and HT22 cells were placed in the lower chamber (effector cells). The SAH model was simulated by oxygenated hemoglobin (40 µM, MilliporeSigma, Burlington, MA, USA) treatment for 24 h.
Frozen brain sections (7 µm) and cultured neurons were both used for
immunofluorescence staining. The slices were treated with 0.3% Triton X-100 for
30 min and 5% donkey serum for 1 h, sequentially. Then, anti-ionized
calcium-binding adapter molecule 1 (Iba-1) (1:200, Cell Signaling Technology,
20825, USA), anti-neuronal nuclear protein (NeuN) (1:200, Abcam, ab177487, UK),
anti-glial fibrillary acidic protein (GFAP) (1:200, Abcam, ab68428, UK),
anti-myeloperoxidase (MPO) (1:200, Santa Cruz Biotechnology, sc-390109, USA), or
anti-CD68 (1:250, Abcam, ab201844, UK) antibody was incubated with the tissue
slices at 4 °C overnight. After washing with phosphate-buffered saline
(PBS), the brain slices were incubated with anti-TREM2 antibody (1:200, Abcam,
ab86491, UK). After washing with PBS–Tween, the slices were incubated with the
proper fluorescently labeled secondary antibody. Subsequently,
4
A One Step TUNEL Assay Kit (KeyGen BioTECH, Nanjing, China) with either brain tissue or cultured cells was used, according to the manufacturer’s instructions.
An annexin V-adenomatous polyposis coli (APC)/7-aminoactinomycin D (7-AAD)
apoptosis detection kit (KeyGen BioTECH) was employed for staining cells, and
flow cytometry was adopted to detect and analyze the cells. The cells were
cultured in 60-mm dishes. After oxygenated hemoglobin (40 µM) treatment for
24 h, the lower HT22 cells were digested and collected with 0.25% EDTA-free
trypsin. Then, the cells were washed twice with PBS (centrifugation at 1000 rpm,
5 min), and 5
Temporal cortical tissues or cultured neurons were collected and lysed in radioimmunoprecipitation assay buffer (Beyotime, Shanghai, China) for 10 min, and then the supernatant was collected after centrifugation at 12,000 g for 15 min. The protein concentrations were determined by using a detergent-compatible protein assay (Beyotime, Shanghai, China).
After separation by 12% sodium dodecyl sulfate–polyacrylamide gel
electrophoresis, the proteins were transferred onto a polyvinylidene difluoride
membrane (Millipore, Billerica, MA, USA). After blocking with 5% nonfat milk for
2 h, the membrane was then serially incubated with primary antibodies (TREM2,
GeneTex, GTX53229, 1:1000; cleaved caspase 3, ImmunoWay, YC0006, 1:1000; cleaved
caspase 1, ImmunoWay, YC0003 1:1000; GAPDH, Proteintech, 60004-1-lg, 1:50,000;
Bcl-2, Abcam, ab182858, 1:2000; Bax, Proteintech, 50599-2-lg, 1:2000; GSDMD-N,
ImmunoWay, TY7991, 1:1000; IL-1
Total RNA was extracted and converted to cDNA, according to the manufacturer’s
guidelines. The primers used are listed in Table 1. PCR was performed in a
20-µL reaction mixture, including 10 µL of 2
| Gene name | Primer sequence |
| Cleaved caspase 3 | Forward: TGGAGGCTGACTTCCTGTATGC |
| Reverse: GAACCACGACCCGTCCTTTGA | |
| Bcl-2 | Forward: GCTACGAGTGGGATGCTGGAGA |
| Reverse: GGTTGCTCTCAGGCTGGAAGGA | |
| Bax | Forward: CCAGGATGCGTCCACCAAGAAG |
| Reverse: CCGTGTCCACGTCAGCAATCAT | |
| Cleaved caspase 1 | Forward: GGACTGACTGGGACCCTCAAGT |
| Reverse: GGCAAGACGTGTACGAGTGGTT | |
| GSDMD-N | Forward: ACTGAGGTCCACAGCCAAGAGG |
| Reverse: CCACTCGGAATGCCAGGATGCT | |
| IL-1 |
Forward: TCGCAGCAGCACATCAACAAGA |
| Reverse: CCACGGGAAAGACACAGGTAGC | |
| GAPDH | Forward: AAGGTCGGTGTGAACGGATT |
| Reverse: TGAGTGGAGTCATACTGGAACAT |
All experimental results are shown as the mean
A total of 251 mice were used for this study, including 209 mice that were exposed to SAH. All mice in the sham group survived. The total mortality rate was 17.70% (37/209). SAH was successfully induced in 156 mice. Sixteen mice were excluded from this study due to a lack of hemorrhage. The typical brains of SAH and sham mice are shown in Fig. 1a.
 Fig. 1.
Fig. 1.Increased basal cortex TREM2 expression after subarachnoid hemorrhage (SAH). Male
C57BL/6J mice were assigned into the sham or SAH group (n = 6). The SAH model was
built by endovascular perforation. Brain samples were obtained at the indicated
time points. The protein and mRNA levels of TREM2 were determined by western blot
and RT-qPCR, respectively. (a) Typical mouse brains of sham and SAH mice post
SAH. (b) Western blot result of TREM2 protein in the basal cortex at 72 h post
SAH. GAPDH was used as the loading control. (c) Quantitative analysis of the
western blot results. (d) RT-qPCR results of TREM2 mRNA at the indicated time
points. (e) Immunofluorescence staining of TREM2 (green) in the basal cortex at
72 h post SAH, while Iba1 (microglia), NeuN (neuron), and GFAP (astrocyte) are
shown in red. The cell nuclei stained with DAPI are shown in blue. ****p
By performing western blot and RT-qPCR assays, we determined the protein and mRNA levels of TREM2. Compared to the levels in the sham mice, the results indicated that the TREM2 protein and mRNA levels were significantly increased in the SAH mice at 2 d after SAH, peaked at 3 d, and decreased at 5 d (Fig. 1b–d). Co-immunostaining of TREM2 with Iba1, GFAP, or NeuN showed that TREM2 was mainly expressed in the microglia (Iba1) after SAH (Fig. 1e). Only trace TREM2 immunostaining signals were observed in astrocytes (GFAP) and neurons (NeuN) (Fig. 1e). These results indicate that TREM2 plays a role in SAH, which may be mediated by microglia.
Since TREM2 exerted an immediate response in SAH, we wondered whether the change in the TREM2 level after SAH was protective or destructive. Thus, we manipulated the expression level of TREM2 in the mouse brain by shRNA knockdown and overexpression intraventricularly. First, we evaluated the neurobehavior of the mice. The Garcia Neuroscores of the SAH mice were found to be lower than those of the sham mice (Fig. 2a). In addition, the sh-TREM2 mice were shown to have a lower score after SAH compared to the mice that only underwent SAH. These results indicate that TREM2 may play a protective role after SAH.
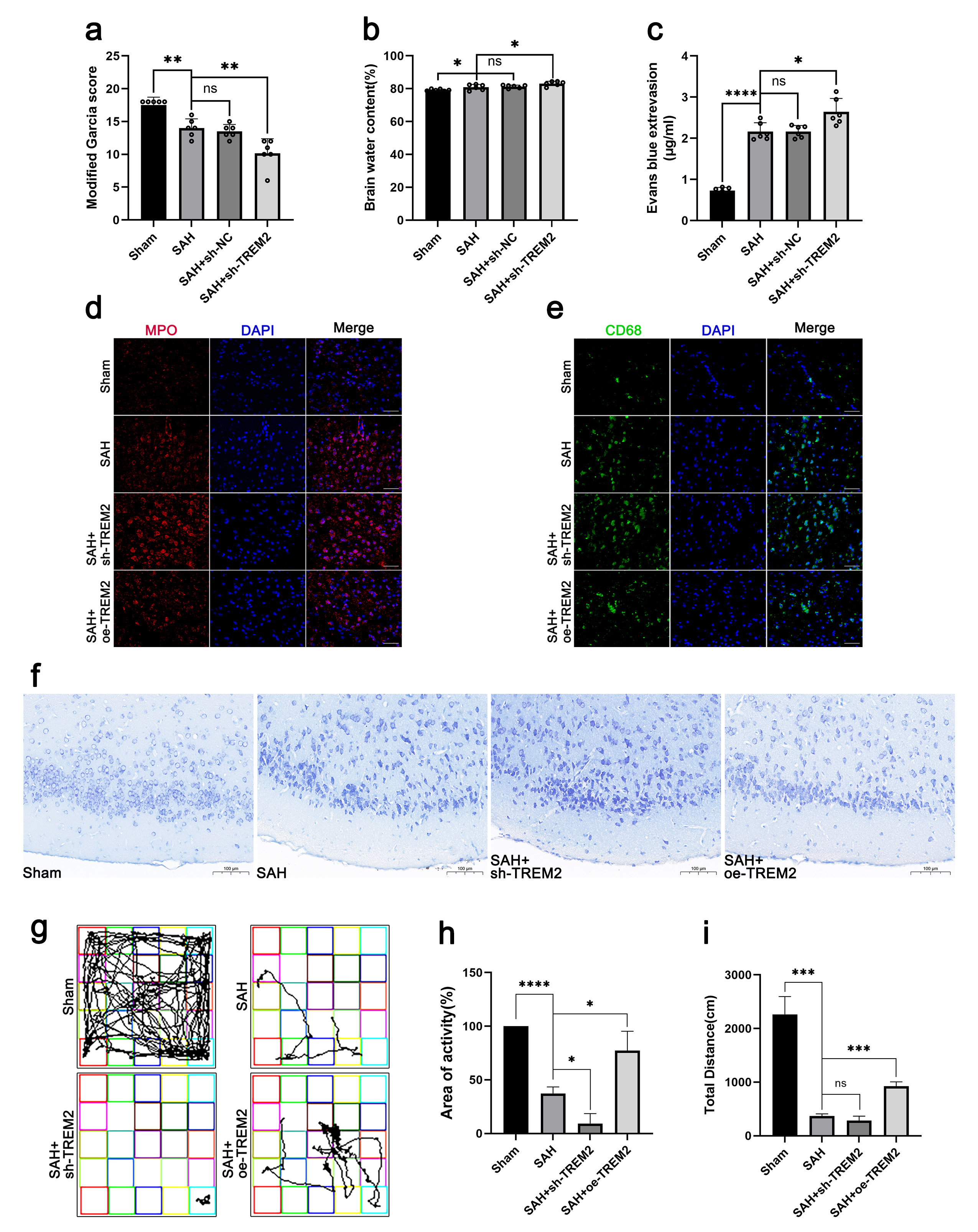 Fig. 2.
Fig. 2.TREM2 overexpression rescued impaired neural function.
C57BL/6J mice were randomly divided into five groups: sham, SAH, SAH nc-TREM2,
SAH sh-TREM2, and SAH oe-TREM2. Seven days before SAH model construction,
lentivirus (8 µL) was injected into the right ventricle using an
automatic brain stereotaxic apparatus. Neurobehavior assessment and the open
field test were performed at 72 h after SAH. After sacrifice, the brain water
content of the mice was evaluated. Evans blue (EB) staining, Nissl staining, and
immunofluorescence staining of MPO and CD68 were performed. (a) Neurobehavior
score. (b) Brain water content. (c) EB staining of the blood–brain barrier.
(d,e) Immunofluorescence staining of MPO and CD68. Scale bar = 50 µm. (f)
Nissl staining. Scale bar = 100 µm. (g–i) Open field test. Typical
pictures of the route (g), total traveled distance (h), and percentage of area of
activity (i) are shown. ns, no significance, *p
Afterwards, the brain water content of all mice was also determined (Fig. 2b).
The brain water content of the SAH mice was significantly increased, while
sh-TREM2 treatment aggravated it. By immunohistochemistry and an EB diffusion
assay, we found that the blood–brain barrier (BBB) permeability (Fig. 2c),
neuronal degeneration (Fig. 2f), neutrophil infiltration (MPO
In the open field test (Fig. 2g–i), the SAH mice travelled significantly less than the sham mice, while the sh-TREM2 mice were shown to be less active than the SAH mice. However, the oe-TREM2 mice exhibited a better performance in the open field test, showing a greater total travelling distance and a higher activity compared to those of the SAH mice. Therefore, TREM2 was demonstrated to rescue the impaired neural function in the SAH mice.
TREM2 was shown to suspend inflammation. Besides, cell apoptosis and pyroptosis
also were shown to follow brain damage after SAH. Here, to clarify the regulatory
role of TREM2 in SAH, we further detected cell apoptosis and pyroptosis in the
brain. As shown in Fig. 3a–c, the levels of cleaved caspase 3, Bax, cleaved
caspase 1, GSDMD-N, and IL-1
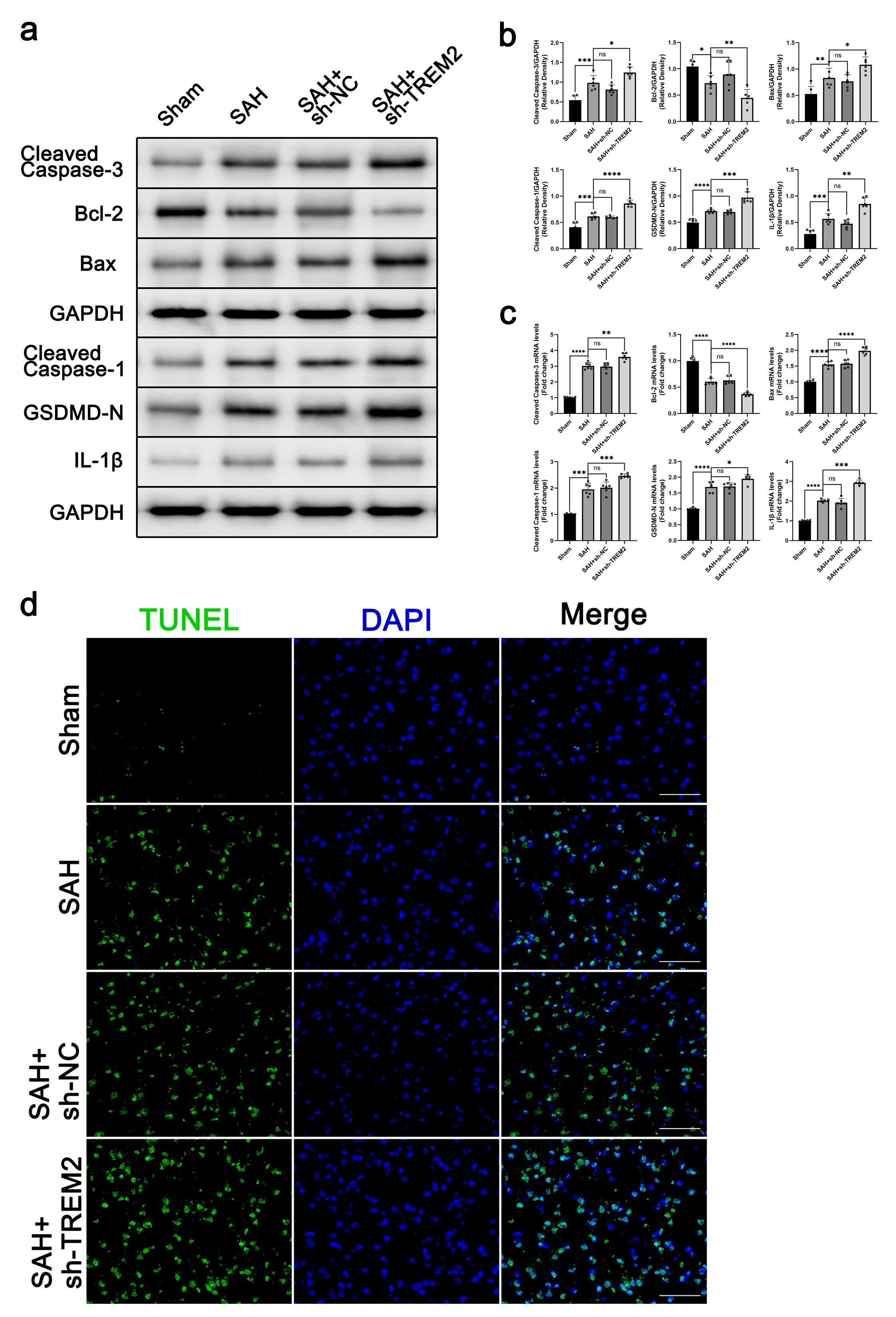 Fig. 3.
Fig. 3.TREM2 knockdown worsened inflammation and cell death in SAH
mice. C57BL/6J mice were randomly divided into four groups: Sham, SAH,
SAH+sh-NC, and SAH+sh-TREM2. At 72 h post SAH, the brain samples were prepared
for western blot and RT-qPCR assays for cleaved caspase 3, Bcl-2, Bax, cleaved
caspase 1, GSDMD-N, and IL-1
To confirm the protective effect of TREM2 on neurons, we adopted an
in-vitro SAH cell model for further investigation. By lentiviral
transfection, sh-TREM2 (by shRNA) and oe-TREM2 (by overexpression of TREM2)
stable BV-2 cell lines were constructed. After the coculture of BV-2 microglia
and HT22 neuronal cells, cell apoptosis and inflammation were detected in HT22
neurons. As shown in Fig. 4a–c, the levels of cleaved caspase 3, Bax, cleaved
caspase 1, GSDMD-N, and IL-1
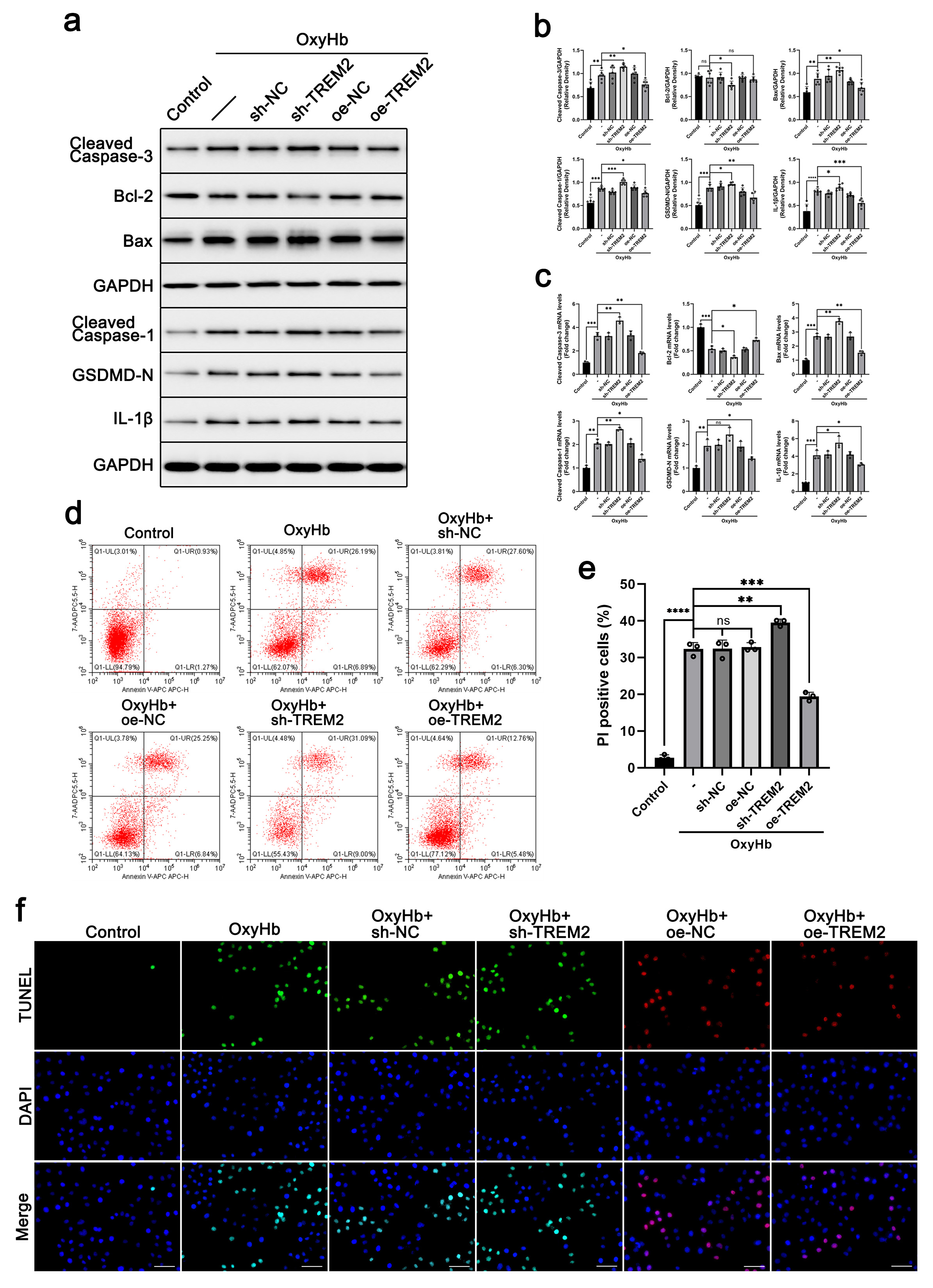 Fig. 4.
Fig. 4.TREM2 showed a neuroprotective effect in an
in-vitro SAH cell model. sh-TREM2 (by shRNA) and oe-TREM2 (by
overexpression of TREM2) stable BV-2 cell lines were constructed by lentiviral
transfection. BV-2 and HT22 neuronal cells were cocultured, and the SAH model was
simulated by adding 40 µM oxygenated hemoglobin for 24 h. The protein
levels of cleaved caspase 3, Bcl-2, Bax, cleaved caspase 1, GSDMD-N, and
IL-1
In the brain, neurons are responsible for neuronal functions, while microglia serve an auxiliary role. Microglia can exert either a protective or deleterious impact upon neurons, depending on the time and conditions [31]. In SAH, neuroinflammation caused by microglial activation is tightly connected to the secondary injury. A therapeutic strategy targeting neuroinflammation has been reported to reduce neuronal damage and to improve neurological dysfunction [32]. However, until now, the underlying mechanism has not been elucidated. Recently, evidence has shown that TREM2, a specific anti-inflammatory receptor in the microglia, plays a pivotal and protective role in modulating neuroinflammation [33]. Here, we explored the protective effect of TREM2 in the early secondary brain damage after SAH. By using the SAH mouse model, we determined the expression level of TREM2 after SAH. By comparing the behavioral and biochemical results from the mice with TREM2 knockdown (sh-TREM2) and overexpression (oe-TREM2), we found that the cognitive performance of the mice with oe-TREM2 after SAH was better than that in those without TREM2 alteration after SAH, while the cognitive performance of the mice with sh-TREM2 after SAH was worse than that in those without TREM2 alteration after SAH. The water brain content, BBB permeability, and programmed cell death (including apoptosis and pyroptosis) were all deteriorated in the sh-TREM2 mice compared with those in the mice without TREM2 alteration. This result was confirmed by an in-vitro SAH cell model. The overexpression of TREM2 in microglia can decrease the cell population in apoptosis in cocultured SAH neurons, while downregulated TREM2 expression in microglia can increase the population of cocultured SAH neurons in apoptosis. Both apoptosis and pyroptosis were manipulated by the alteration of TREM2 expression. Our results provide more evidence of the protective function of TREM2 in brain injury induced by SAH and especially underline its role in programmed cell death, including apoptosis and pyroptosis.
The TREM2 pathway has been confirmed to play roles in several diseases. For
example, variants of TREM2 have been found to be associated with
neurodegeneration, such as in Alzheimer’s disease [33]. In addition, TREM2 has
been demonstrated to be involved in the microglial activation against amyloid
plaques, which are the main characteristic of Alzheimer’s disease. Loss of TREM2
function also has been shown to reduce the microglial responses to amyloid
plaques, which become more toxic [34]. Evidence indicates that the overexpression
of TREM2 can decrease the formation of amyloid plaque and alleviate cognitive
deficits [35], and this process is mediated by microglia. The sTREM2 level in the
CSF has been suggested to be used as a biomarker of Alzheimer’s disease [36]. In
cancer, TREM2 is normally overexpressed and used as a marker for macrophages and
monocytes [37]. In hepatocellular carcinoma, the disruption of TREM2 expression
promotes tumor development and exacerbates inflammation in the liver. In the
intestine, TREM2 is expressed in human monocyte dendritic cells and is limited to
inflamed sites, contributing to the pathogenesis of inflammatory bowel diseases
[38]. Besides, in many types of strokes, such as ischemic stroke, TREM2 can
reduce inflammation via the Toll-like receptor signaling pathway, thus
promoting the migration, survival, and regeneration of microglia [39]. Previous
studies and our present results all showed that the expression of TREM2 responded
quickly to SAH, reaching a peak at 24–72 h after brain injury [17]. The
pathophysiological changes following SAH, including erythrocyte leakage to the
subarachnoid space and resident microglia/macrophage (Mi/M
Since the neuroprotective function of TREM2 in SAH and other related disorders
has been confirmed in many studies, TREM2 is thought to be a good therapeutic
target. However, TREM2 displays a distinct role in different disorders and even
at different stages of one disease; therefore, the modulation of TREM2 should be
based on clarification of the related mechanism. For example, coupling TREM2 and
apolipoprotein to promote phagocytosis of impaired neurons or clearance of
amyloid plaque by microglia may be used to treat Alzheimer’s disease [42, 43].
Regulating the release rate of sTREM2 is another strategy to cope with the
pathology of Alzheimer’s disease [44]. In stroke, whether TREM2 is involved in
erythrocyte and metabolite clearance still needs to be investigated. Besides,
since both TREM2 overexpression and activation can enhance the phagocytic ability
of Mi/M
In conclusion, by constructing in-vivo and in-vitro SAH models, the neuroprotective mechanisms of TREM2 after SAH were investigated. The knockdown of TREM2 in the mouse brain aggravated cognitive impairment, BBB permeability, and cell death (apoptosis and pyroptosis). Moreover, in cocultured microglia and neurons, the overexpression of TREM2 in microglia decreased the cell apoptosis and pyroptosis of neurons after SAH. Thus, TREM2 alleviates secondary brain injury through attenuating cell death in both mice and cultured neurons with SAH, making TREM2 a promising therapeutic target for SAH.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
JL and ZZ performed the experiments and drafted the manuscript. JL, ZZ, MZ, SL, XZho, and ZL participated in the experimental design and conceived. MQ and YH designed the feeding protocol, helped to feed the mice, and coordinated the study. BY and FQ performed the neurobehavioral studies as well as participated in the sample collection and staining experiments. SL, XZho, XZha, and ZL participated in data analysis and reviewed the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Animal handling and all of the related experimental procedures were carried out according to the National Institutes of Health guidelines and approved by the Animal Ethics Review Committee of Wannan Medical College (approval number: WNMC-AWE-2023293).
Not applicable.
This work was supported by Shandong Provincial Third Hospital [3450019009], the Anhui Provincial Department of Education Natural Science Major Projecti [2023AH040240], and the Professional Science Research Project of the First Affiliated Hospital of Wannan Medical College [YR202004].
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.