
- Academic Editor
The abnormal intermediate glucose metabolic pathways induced by elevated intracellular glucose levels during hyperglycemia often establish the metabolic abnormality that leads to cellular and structural changes in development and to progression of diabetic pathologies. Glucose toxicity generally refers to the hyperglycemia-induced irreversible cellular dysfunctions over time. These irreversible cellular dysfunctions in diabetic nephropathy include: (1) inflammatory responses, (2) mesangial expansion, and (3) podocyte dysfunction. Using these three cellular events in diabetic nephropathy as examples of glucose toxicity in the diabetic complications, this review focuses on: (1) the molecular and cellular mechanisms associated with the hexosamine biosynthetic pathway that underly glucose toxicity; and (2) the potential therapeutic tools to inhibit hyperglycemia induced pathologies. We propose novel therapeutic strategies that directly shunts intracellular glucose buildup under hyperglycemia by taking advantage of intracellular glucose metabolic pathways to dampen it by normal synthesis and secretion of hyaluronan, and/or by intracellular chondroitin sulfate synthesis and secretion. This could be a useful way to detoxify the glucose toxicity in hyperglycemic dividing cells, which could mitigate the hyperglycemia induced pathologies in diabetes.
Hyperglycemia is a main feature of diabetes, and one third of the adult population has a higher blood glucose concentration that can be classified as pre-diabetic [1]. Under hyperglycemia, the extensive and chronic inflammatory responses have been considered as one of the early events that leads to development of diabetic complications. In diabetic nephropathy, hyperglycemia-induced chronic inflammation, mesangial expansion, and podocyte dysfunction have been considered as the events that are critical for the development and progression of the disease. However, the underlying mechanisms are still not clear. Hyperglycemia induces an increase in intracellular glucose that activates intermediary metabolic pathways, including polyol, advanced glycation end product (AGE), protein kinase C (PKC), and hexosamine biosynthesis [2, 3], which leads to glucose toxicity [4, 5]. The pathogenesis of diabetic nephropathy and the abnormality of metabolic pathways induced by hyperglycemia have been extensively investigated and well documented in previous reviews [5, 6, 7, 8, 9, 10, 11]. This paper focuses on the hexosamine biosynthetic pathway (HBP) to identify: (1) molecular and cellular mechanisms associated with HBP that underly diabetic complications such as diabetic nephropathy, and (2) potential therapeutic tools to inhibit this disease.
HBP is a glucose metabolic pathway through which ~3% of intracellular glucose is used. This pathway starts from fructose-6-phosphate (F-6-P) to produce the main product, uridine diphosphate- N-acetylglucosamine (UDP-GlcNAc) by a series of enzymatic reactions. The first step in this pathway is the rate limiting reaction to convert F-6-P to glucosamine 6-phosphate (glcN-6-P) catalyzed by the glutamine:fructose-6-phosphate amidotransferase (GFAT) [12, 13, 14]. Once formed, glcN-6-P is subsequently metabolized to UDP-GlcNAc by several enzymatic reactions. Therefore, under hyperglycemia, increased intracellular glucose elevates the UDP-GlcNAc production, which is indicated by increases in hyaluronan synthesis [15] and O-Glycosylation [4, 5], in which UDP-GlcNAc is served as a rapidly utilized substrate. The GFAT activity determines the extent of intracellular glucose utilized by this pathway, and its activity is highly regulated by: (1) the F-6-P level; (2) the negative feedback of UDP-GlcNAc; and (3) GFAT expression and posttranslational modification [12]. GFAT is expressed in diabetic glomeruli with a half-life of less than one hour, and is absent in the normal glomeruli [12, 16]. Further, the expression of GFAT can be regulated by epidermal growth factor (EGF), glucose and glucosamine, and by Sp1 GC-boxes [17]. Interestingly, the CCAAT/enhancer-binding proteins (C/EBP) within the cyclin D3 complex can activate the CCAAT-boxes in the GFAT promoter [17, 18]. Therefore, our studies [19], provide evidence that high glucose directly induces GFAT expression by showing that the complex of C/EBP and cyclin D3 is increased under hyperglycemia.
Hyperglycemia in diabetes activates the HBP, an important consequence of elevated intracellular glucose that contributes to the genesis and progression of diabetic nephropathy [6, 12, 13, 20]. Preclinical and clinical studies have shown: (1) increased numbers of activated inflammatory monocytes/macrophages [21, 22, 23] (our study [24]); (2) an expanded mesangial matrix; and (3) impaired function of podocytes [25, 26, 27]. Euglycemic control in diabetes minimally reverses the diabetic complications [28]. A previous study [6] and our studies [19, 29, 30, 31, 32] indicate that hyperglycemia-induced UDP-GlcNAc production by the activated HBP is the central mechanism involved (Fig. 1, Ref. [6, 15, 33]) as shown in diabetic animal models and human patients [34, 35]. This abnormal increase in UDP-GlcNAc production could have essential roles in mediating inflammatory responses, mesangial expansion, and podocyte dysfunction, which are poorly understood [25, 26, 27].
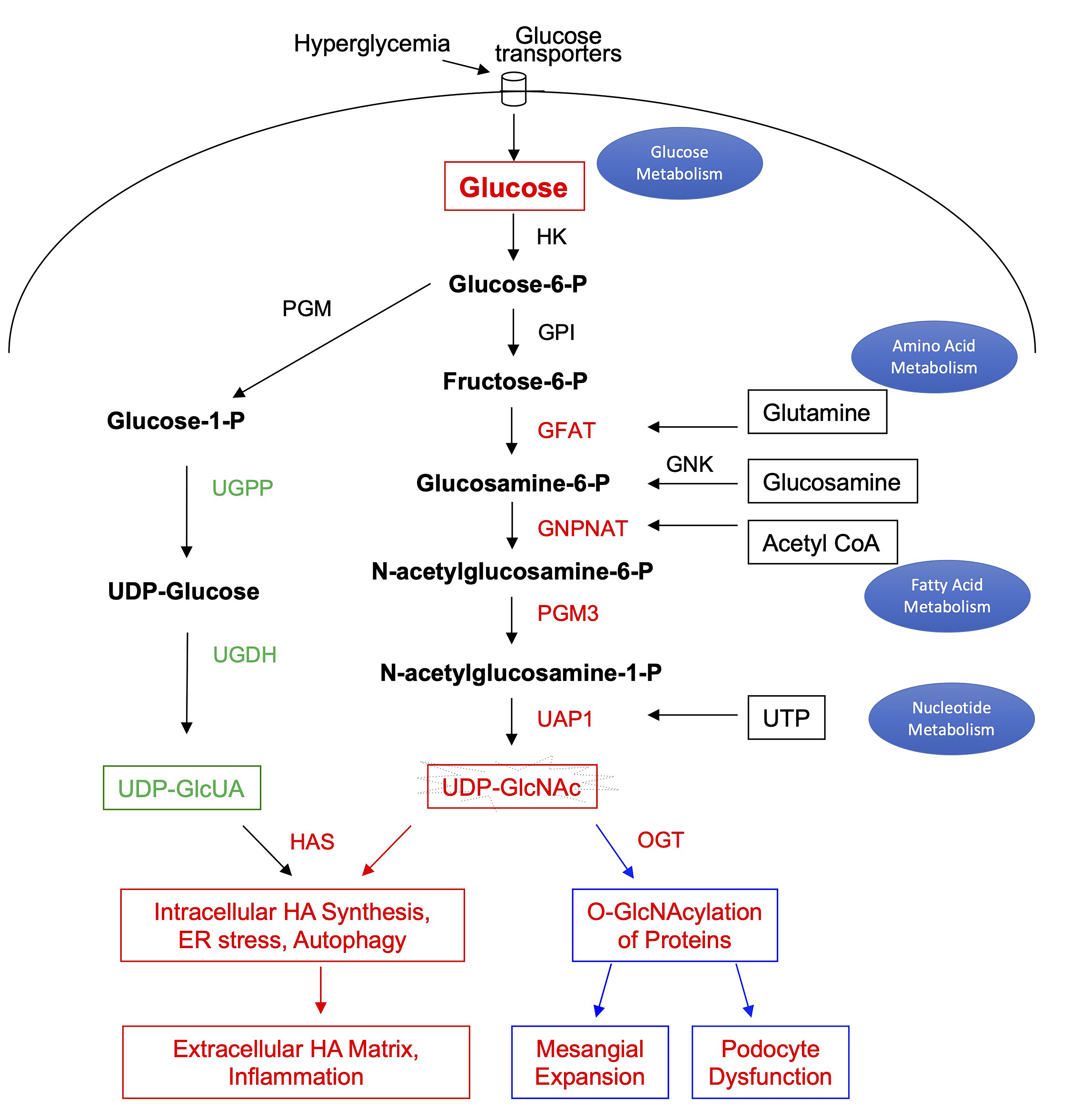 Fig. 1.
Fig. 1.The HBP modulates hyperglycemia induced inflammatory responses, mesangial expansion and podocyte dysfunction through induction of HA synthesis and protein O-linked-N-acetylglucosaminylation (O-GlcNAcylation) inside of cells [6, 15, 33]. (1) Uronic acid synthetic pathway: uridine diphosphate-glucuronic acid (UDP-GlUcA) synthesis begins with glucose-6-phosphate (Glu-6-P) and ends with oxidation of uridine diphosphate-glucose (UDP-GlUc) catalyzed by UDP-glucose dehydrogenase (UGDH); and (2) Hexosamine biosynthetic pathway: UDP-GlcNAc is produced from fructose-6-phosphate (Fru-6-P), and the first reaction of this pathway, the production of D-glucosamine-6-phosphate, is considered the rate-limiting step. Under hyperglycemia, UDP-GlcA and UDP-GlcNAc are used to synthesize hyaluronan in the intracellular compartments, including the ER that induces ER stress, autophagy and subsequent inflammatory responses. On the other hand, elevated UDP-GlcNAc production increases O-GlcNAcylation of proteins that induces mesangial expansion and podocyte dysfunction. HBP, Hexosamine Biosynthetic Pathway; HA, hyaluronan; GlcNAc, N-acetylglucosamine; UDP, uridine diphosphate; ER, endoplasmic reticulum; PGM3, phosphoglucomutase 3; HK, hexokinase; GPI, glucose-6-phosphate isomerase; GFAT, glucosamine fructose-6-phosphate aminotransferase; GNPNAT, glucosamine 6-phosphate N-acetyltransferase; GNK, N-acetylglucosamine kinase; UAP1, UDP-GlcNAc pyrophosphorylase 1; UGPP, UDP-glucose pyrophosphorylase; UDP-GlcUA, UDP-glucuronic acid; HAS, hyaluronan synthase; OGT, O-GlcNAc transferase.
Under hyperglycemia, our studies [19, 29, 30, 31, 36] and others [6, 37] show that increased cytoplasmic UDP-GlcNAc production in dividing cells initiates two responses: (1) activation of HA synthesis inside the dividing cells that induces ER stress/autophagy and subsequently formation of an extracellular monocyte adhesive hyaluronan (HA) matrix, which leads to chronic inflammation; and (2) an increase in cytosolic protein O-GlcNAcylation, which leads to mesangial expansion in the mesangium, foot process effacement, and slit diaphram (SD) dysfunction in podocytes (Fig. 1) [6, 37]. Further, O-GlcNAcylation is required for both the hyaluronan synthase (HAS) expression and activation [38], which provides evidence that the intracellular HA synthesis and protein O-GlcNAcylation can coordinately mediate the glucose toxicity induced by hyperglycemia. We have used the Streptozotocin (STZ) diabetic rat model to explore the in vivo responses [19, 30].
Hyaluronan, central to our studies, is a glycosaminoglycan (GAG) that contains disaccharide repeats of N-acetylglucosamine (GlcNAc) and glucuronic acid (GlcUA) with one negative charge on each GlcUA under physiological pH [15]. HA has a very simple structure that contains more than 1000 disaccharide repeats with a molecular weight of 5–10 MDa. Due to its hydrophilic properties, HA can occupy a large space. Through its binding proteins, HA has important roles in different physiological processes, and critical roles in many pathological reponses mediating disease development and progression [15]. Normally, HA is synthesized by HAS on the plasma membrane of non-dividing cells and secreted into the extracellular environment. Dividing cells in normal glucose remove the glucose transporters from the cell surface and only activate HA synthesis and HA extrusion after division. Once activated on the plasma membrane, HAS synthesizes the HA from cytosolic UDP-GlcNAc and UDP-GlcUA and extrudes the growing (GlcNAc-GlcUA) disaccharide chains through the plasma membrane to form extracellular HA matrices and cell surface coats [15, 29, 32, 33, 39].
However, in hyperglycemic dividing cells, HAS is synthesized in intracellular membranes to decrease the continuous influx of glucose since the cell surface glucose transporters were not removed from the cell surface. Our previous studies on HA [19, 30, 36] show that dividing rat kidney mesangial cells (RMCs) under high glucose activate HAS inside the cells when the cells enter S phase. This activated HAS on the intracellular membranes decreases the intracellular glucose levels by synthesis of the large and polyanionic HA inside the cells and inserts it into ER and golgi. This initiates ER stress and autophagy and then formation of an extracellular monocyte-adhesive HA matrix after division. These studies focused on the HA-based matrix that is synthesized by dividing cells stressed by hyperglycemia, and on its dialogue with associated macrophages, which are derived from circulating monocytes that are recruited into the inflamed tissues. The results provide evidence that this monocyte-adhesive HA matrix and its interactions with the macrophages have a central role in defining the resulting inflammatory pathologies [15, 33].
O-GlcNAcylation, another central cytosolic pathway related to our studies, is a dynamic intracellular protein post-translational modification that cycles on and off serine and/or threonine hydroxyl groups of over 4000 proteins [40]. Cycling of O-GlcNAc is regulated by the concerted actions of nucleocytoplasmic O-GlcNAc transferase (OGT), which adds O-GlcNAc [41], and nucleocytoplasmic glycoside hydrolase O-GlcNAcase (OGA), which removes O-GlcNAc [42]. Cytosolic levels of O-GlcNAc serve as a rheostat to control the intensity of intracellular signaling to apoptosis via nutritional cues from the surrounding environment [40, 43, 44, 45]. Therefore, under hyperglycemia, the excess of nutrients could destroy this delicate balance and result in dysregulation of O-GlcNAcylation and cellular processes, and ultimately induce disease progression [43]. Indeed, O-GlcNAcylation is abnormally and globally increased in the cells and tissues of diabetic animals and humans [34, 35] that causes dysregulation of signaling cascades and transcription levels [46, 47, 48, 49]. O-GlcNAc modifications of cytosolic proteins are increased in human kidney biopsy specimens from diabetic patients [46]. However, little is known about the role of O-GlcNAc in glucose toxicity and diabetes.
Previous studies on O-GlcNAcylation have shown that protein O-GlcNAcylation is a nutrient sensor mediating the pathogenesis of metabolic diseases, including diabetes [6, 40, 50]. Previous studies provided evidence that O-GlcNAc contributes to increased extracellular matrix (ECM) in mesangial cells by altering the expressions of genes and proteins, a hallmark of diabetic nephropathy (DN) in hyperglycemic conditions [6, 37]. These responses would be inhibited by a reduction in O-GlcNAc levels. Further, O-GlcNAc regulates the expression of slit diaphram (SD) specific proteins in podocytes resulting in loss of the SD glomerular size-selective barrier [6, 37]. The proposed mechanism to link O-GlcNAcylation to diabetic podocyte dysfunction has been described previously [6]. This process is initiated by increased O-GlcNAcylation as a result of increased OGT specific activity and decreased OGA specific activity. This imbalance in O-GlcNAc levels leads to alterations of gene expression, protein expression, and/or localization of two essential SD-specific proteins, nephrin and podocin [6]. Alterations in podocin expression directly affect nephrin localization. Further, there is an increase in occluden-type proteins, including junctional adhesion molecule A and zonula occludens-1, that attribute to the SD shift from a modified adherent junction to an increased formation of tight junctions [51]. Thus, decreasing O-GlcNAcylation can likely inhibit the diabetic renal disease-associated phenotype.
Hyperglycemia induced pro-inflammatory responses, mesangial expansion, and
podocyte dysfunction are due to the increased intracellular HA synthesis and the
aberrant O-GlcNAcylation of proteins stimulated by the excessive flux of the
HBP. Our studies [30, 52] and those from others [25, 26, 27] indicate that
pro-inflammatory responses, mesangial expansion, and podocyte dysfunction are
induced by hyperglycemia in the diabetic kidney glomeruli. The abnormal
accumulation of the very large and polyanionic HA within intracellular
compartments (ER, Golgi, transport vesicles) and the enhanced O-GlcNAcylated
intracellular proteins of kidney mesangial cells and podocytes are directly
stimulated by excessive HBP activation (Fig. 1). Thus, these results suggest a
novel strategy to ameliorate inflammatory processes prevalent in the current
extensive diabetic pathologies. Hyperglycemic intracellular glucose induces
abnormal metabolism and has been central in diabetic pathologies. Heparin,
heparin trisaccharide (Hep-Tri) and 4-methylumbelliferyl-
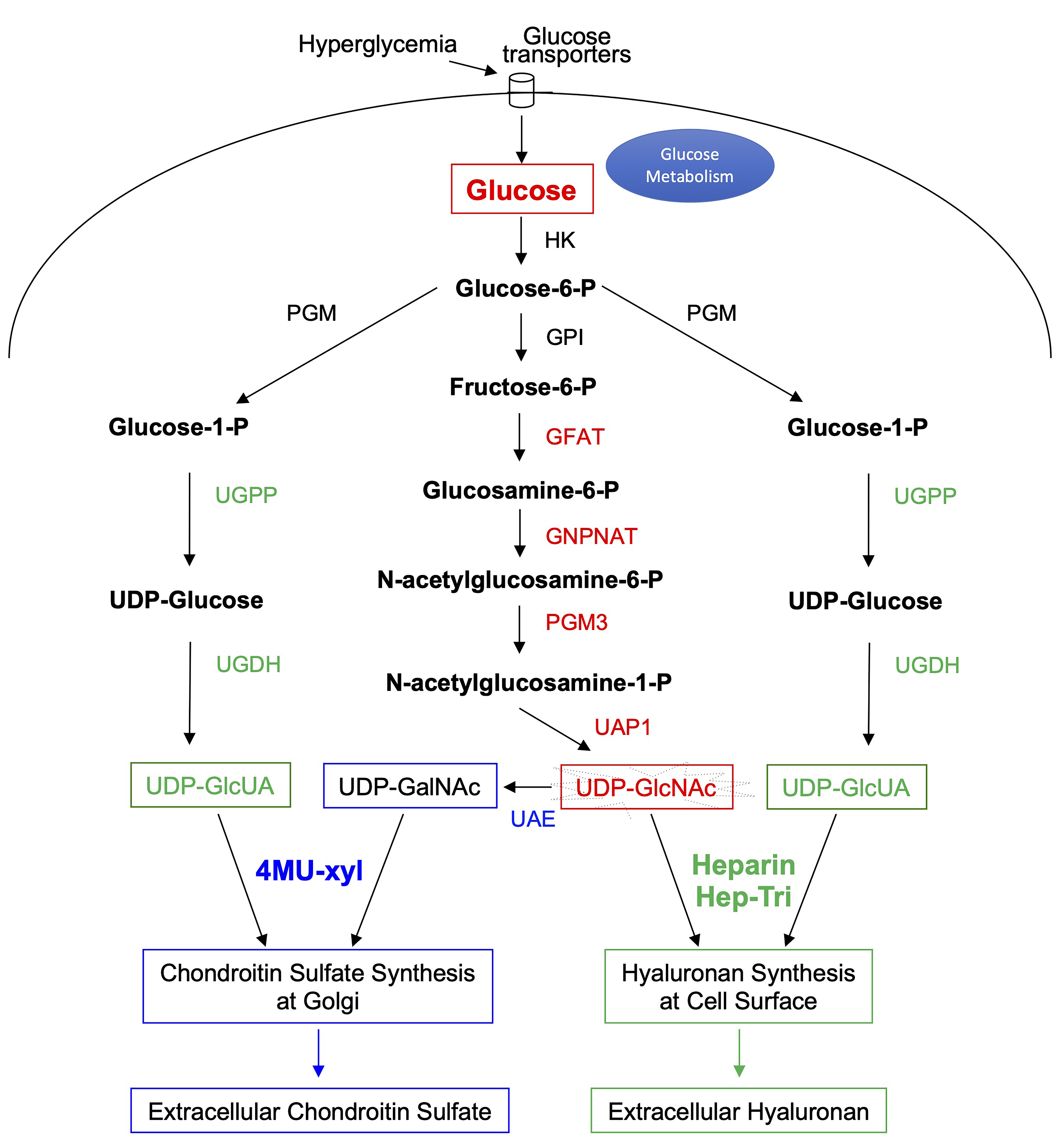 Fig. 2.
Fig. 2.Detoxification of high intracellular glucose by heparin/Hep-Tri
and 4MU-xyl by activation of hexosamine and uronic acid pathways. 4MU-xyl,
4-methylumbelliferyl-
Our novel hypotheses are: (1) that hyperglycemia induces increases in cytosolic glucose metabolites and UDP-sugars in dividing cells that initiates ER stress and autophagy due to intracellular HA synthesis and cytosolic protein O-GlcNAcylation, which induces inflammation, mesangial expansion and podocyte dysfunction; (2) that heparin and its non-reducing trisaccharide Hep-Tri prevent intracellular hyaluronan synthase activation induced by hyperglycemic dividing cells and induces extracellular HA synthesis to form a monocyte-adhesive extracellular HA matrix after completing division, which prevents intracellular glucose stress [30, 53]; and (3) that 4MU-xyl uses cytosolic UDP-GalNAc and UDP-GlcUA substrates to synthesize chondroitin sulfate in Golgi, which prevents accumulation of cytosolic glucose via the HBP and uronic acid pathways [24, 32, 33, 55].
Future studies should pursue three goals: (1) determine the underlying mechanisms for how the HBP activation induced by hyperglycemia on kidney cells leads to the 3 abnormal responses, and identify ways to prevent them; (2) identify the impacts of the interplay between the HBP activation, intracellular HA accumulation and O-GlcNAcylation of cytosolic proteins that mediate these 3 early responses (Fig. 1); and (3) test the therapeutic potentials of heparin, Hep-Tri and 4MU-xyl treatments for these hyperglycemia-induced abnormal responses (Fig. 2). These proposed studies are based on our finding [53] that the Hep-Tri, that does not have anticoagulant activities [56, 57], inhibits the hyperglycemia-induced stress responses, and that 4MU-xyl, which has been used previously in preclinical studies [58, 59], also inhibits the stress functions by a mechanism different from Hep-Tri.
The intracellular HA and increased O-GlcNAcylation are two key mediators of glucotoxicity in the diabetic kidney, and future studies can use the regulators of HA synthesis and O-GlcNAcylation listed in Table 1 to dissect the pathways mediating intracellular HA synthesis and aberrant O-GlcNAcylation. These studies will provide critical information regarding molecular mechanisms in DN and identify potential therapeutic tools. 4MU-xyl increases CS synthesis almost 10-fold in RMCs [30, 55]. This drives the substrates (UDP-GlcUA and UDP-GalNAc) from the cytoplasm into the Golgi through antiporters [33], which blocks intracellular HA synthesis and autophagy, thereby moderating synthesis of the HA matrix [30]. The aglycone 4-methylumbelliferone (4MU) of 4MU-xyl also inhibits HA synthesis by depleting UDP-GlcUA to form 4MU-glucuronides [58], but it has no effect on the production of UDP-GlcNAc via the HBP. Further, Ac4SGlcNAc, an inhibitor of OGT, reduces O-GlcNAcylation, and Thiamet-G (TMG), a highly specific inhibitor of OGA, prevents removal of O-GlcNAc from proteins [50]. Thus, cells dividing in hyperglycemic medium alone, or with these reagents (Table 1) will have distinctly different outcomes, and must have important differences in the signaling pathways involved.
| UDP-GlcNAc | O-GlcNAc | Intracellular HA | ER stress Autophagy | Adhesive HA Matrix | |
| Hyperglycemic | + | + | + | + | + |
| Plus heparin | - | - | - | - | + |
| Plus Hep-Tri | - | - | - | - | + |
| Plus 4MU-xyl | - | - | - | - | - |
| Plus 4MU | + | + | - | - | - |
| TMG | + | + | + | + | + |
| Ac |
+ | - | + | + | + |
+, means increase; -, means decrease. 4MU, 4-methylumbelliferone; TMG, Thiamet-G.
Previous studies have shown the critical role of the HBP in inducing glucose toxicity under hyperglycemia by the increase in O-GlcNAcylation. Therefore, we do expect the proposed treatment could relieve “glucose toxicity”. However, given the complexity of hyperglycemia induced metabolic responses in the diabetic complications [10, 11], the future studies might not show a complete relief of the glucose toxicity. Nevertheless, these proposed studies will provide new insights regarding the potential roles: (1) of the HBP in treatment of diabetic pathologies; and (2) of heparin, Hep-Tri and 4MU-xyl in sustaining renal function by diverting intracellular glucose and O-GlcNAcylation stress to the synthesis of extracellular glycosaminoglycans. These responses would help to develop new strategies to mitigate diabetic complications.
Hyperglycemia induces an influx of glucose into cells that leads to increased glucose levels inside the cells. This buildup of intracellular glucose activates multiple intermediate glucose metabolism pathways that initiate the adverse effects of cells and pathogenesis of diabetic complications. Conventional thinking is to develop inhibitors or blockers to prevent the high intracellular glucose levels that induced the responses and hope that these kinds of reagents could inhibit the hyperglycemia induced pathogenesis. Unfortunately, these therapeutic treatments may inhibit high glucose induced abnormal activations of one or two of the glucose intermediate metabolic pathways [8, 9, 10, 11]. However, they would not lessen the intracellular glucose buildup induced by hyperglycemia. Thus, using an inhibitory strategy might not inhibit the hyperglycemia-induced pathologies in diabetes.
This paper provides a new therapeutic strategy to deal with the adverse glucose buildup inside of cells under hyperglycemia. Instead of blocking glucose shunts into the intermediate glucose metabolic pathways, we propose to use the treatments of heparin, Hep-Tri and 4MU-xyl, to flow the accumulated intracellular glucose through the hexosamine biosynthetic and uronic acid pathways to yield the metabolites that are then used to synthesize HA on the cell surface to form the extracellular HA matrix or to synthesize the chondroitin sulfate in the golgi that is then being secreted into extracellular compartments. These novel therapeutic strategies could directly inhibit intracellular glucose buildup by taking advantage of intracellular glucose metabolic pathways to dampen it by normal synthesis and secretion of HA and/or by intracellular chondroitin sulfate synthesis and secretion. This could be a useful way to detoxify the glucose toxicity in hyperglycemic dividing cells, which could mitigate the hyperglycemia induced pathology in diabetes.
AGE, advanced glycation end product; 4MU, 4-methylumbelliferone; 4MU-xyl,
4-methylumbelliferyl-
AJW, AW and VH designed the review plan. AJW, AW and VH collected and analyzed the literatures. All authors prepared and wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have agreed to be accountable for all aspects of the work.
Not applicable.
An abstract was presented at the 2022 Annual Meeting of the Society for Glycobiology.
This work was supported by National Institutes of Health Grant P01HL107147 (Vincent Hascall Project 1).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.