
- Academic Editor
†These authors contributed equally.
Background: With the recent evolution of multidrug-resistant strains,
the genetic characteristics of foodborne Salmonella enterica serovar
Enteritidis and clinical isolates have changed. ST11 is now the most common
genotype associated with S. Enteritidis isolates. Methods: A
total of 83 strains of S. Enteritidis were collected at the General
Hospital of the People’s Liberation Army. Of these, 37 were from aseptic sites in
patients, 11 were from the feces of patients with diarrhea, and the remaining 35
were of chicken-origin. The minimum inhibitory concentration of S.
Enteritidis was determined by the broth microdilution method. Genomic DNA was
extracted using the QiAamp DNA Mini Kit, and whole-genome sequencing (WGS) was
performed using an Illumina X-ten platform. Prokka was used for gene prediction
and annotation, and bioinformatic analysis tools included Resfinder, ISFinder,
Virulence Factor Database, and PlasmidFinder. IQ-TREE was used to build a maximum
likelihood phylogenetic tree. The phylogenetic relationship and distribution of
resistance genes was displayed using iTOL. Comparative population genomics was
used to analyze the phenotypes and genetic characteristics of antibiotic
resistance in clinical and chicken-origin isolates of S. Enteritidis.
Results: The chicken-origin S. Enteritidis isolates were more
resistant to antibiotics than clinical isolates, and had a broader antibiotic
resistance spectrum and higher antibiotic resistance rate. A higher prevalence of
antibiotic-resistance genes was observed in chicken-origin S.
Enteritidis compared to clinical isolates, along with distinct patterns in the
contextual characteristics of these genes. Notably, genes such as
bla
Salmonella is one of four key causes of diarrheal diseases worldwide [1]. Most cases of salmonellosis are mild, but they can sometimes be life-threatening [2]. The severity of the disease depends on host factors and on the serotype of Salmonella [3]. Salmonella enterica serovar Enteritidis (S. Enteritidis), a non-typhoid Salmonella, causes substantial morbidity and mortality in humans and animals, and is therefore a major concern for public health [4]. S. Enteritidis usually causes acute diarrheal disease after entering the digestive tract with contaminated food. However, it has been widely reported that infection with multidrug-resistant (MDR) S. Enteritidis can lead to non-effective control of clinical infection, resulting in extra-intestinal infections such as bacteremia, endocarditis, meningitis, etc. The prevalence of MDR Salmonella isolates has increased over the last few decades, and antimicrobial-resistant Salmonella isolates have been associated with an increased rate of hospitalization [5, 6]. The World Health Organization (WHO) has declared that antibiotic-resistant S. Enteritidis is a critical-priority bacterium [7].
Since the discovery of penicillin, antibiotics have gradually been used more
widely to treat various bacterial diseases in humans and animals, and to promote
weight gain in animals [8]. However, the serious negative effects and
consequences of antibiotic abuse are now reflected in their residual accumulation
in livestock and poultry. These cause public harm through the emergence of
antibiotic resistance and “super bacteria” in food that threaten human health
[9, 10]. Currently, the fluoroquinolone ciprofloxacin and the third-generation
cephalosporin ceftriaxone are the recommended antibiotics for treating patients
who are at risk of invasive S. Enteritidis infection, with the macrolide
azithromycin as a possible alternative [11]. Most of the severe antibiotic
resistant S. Enteritidis strains are susceptible to carbapenems, making
them the last line of defense in the treatment of Salmonella infections.
However, strains that are resistant to these antibiotics have now emerged. For
example, Abdel-Kader et al. [12] reported the sale of chicken in retail
shops that carried bla
The level of S. Enteritidis resistance is related to the use of antibiotics in humans and animals, geographical differences in epidemiology, and serotypes [15]. ST11 is the most common genotype associated with S. Enteritidis isolates from humans and food animals across the world [16]. The goal of this study was therefore to utilize bioinformatics techniques to clarify the role of horizontal gene transfer in shaping the genome-wide evolution of MDR Salmonella. This should help to reveal the underlying mechanisms that facilitate the swift dissemination of antibiotic resistance genes. For this study, a total of 83 S. Enteritidis strains were collected from different regions in China, including chicken-origin isolates and clinical isolates. We evaluated the prevalence and mechanisms of S. Enteritidis resistance from the different sources by using phenotypic susceptibility data and whole-genome sequencing (WGS) analysis.
A total of 83 S. Enteritidis strains were collected. Clinical isolates originated from samples collected by the clinical laboratory of the First Medical Center, Chinese People’s Liberation Army (PLA) General Hospital, Beijing, China. All strains were isolated from different patients. Thirty-seven strains were isolated from aseptic sites in the human body that were devoid of local inflammation and damage, and were unconnected to the respiratory system, digestive system, urogenital system, or other external environments. These included locations such as venous blood, joint cavities, and cerebrospinal fluid. A further 11 strains were isolated from the feces of diarrheal patients. The other 35 isolates were of chicken-origin obtained from six provinces in China and cultured by the Chinese Institute for Food and Drug Control. The strains were isolated and cultured according to the National Clinical Laboratory Operation Procedures. The genus was identified by a VITEK MS RUO system v3.4 (BioMérieux, Lyon, France), and the serotype by the Salmonella serum agglutination test (Lot: 20210101, Statens Serum Institut, Copenhagen, Denmark).
Genomic DNA was extracted using the QiAamp DNA Mini Kit (Lot: 172030683, Qiagen, Dusseldorf, North Rhine-Westphalia, Germany) and then sequenced using an Illumina X-ten platform to generate 150-bp paired-end reads from a library with an average insert size of 500 bp. Raw reads were first filtered by fastQC v0.11.9 (Babraham Institute, Babraham, Cambridgeshire, England) to remove low-quality reads, and then assembled using SPAdes v3.13 (Saint Petersburg Academic University, Saint Petersburg, Leningrad Oblast, Russia) using the default parameters [17, 18]. The assembled genomes were evaluated by Quast v5.0.2 (Saint Petersburg State University, Saint Petersburg, Leningrad Oblast, Russia). Gene prediction was performed with Prokka v1.14.6 (University of Melbourne, Melbourne, Victoria, Australia), and BLAST v2.2.18 (National Center for Biotechnology Information, Bethesda, MD, USA) was used to compare the virulence genes carried by the strains with the virulence gene database VFDB. The parameters were set to a minimum coverage of 60% and a minimum similarity of 80%. The ResFinder v4.1.11 (Technical University of Denmark, Copenhagen, Denmark) was used to assemble strain sequences related to antibiotic resistance gene screening. The parameters were set to a minimum coverage of 60% and minimum similarity of 80%. The evolutionary tree was constructed with WGS single nucleotide polymorphism (wgSNP) data. MUMmer v3.23 (University of Maryland, College Park, MD, USA) was used to find SNPs in all strains and to merge these. To improve the overall quality of data, a random strain was selected as the reference sequence (FC15971). SNPs less than 5 bp away and those carrying unspecified nucleotides (“N”) were removed, and an evolutionary tree for wgSNP sequences was constructed using fasttree [19, 20, 21, 22, 23]. Antibiotic resistance genes (ARGs) were identified based on the best alignment with the ResFinder database, with thresholds of 90% DNA sequence identity and minimum coverage of 80%. A multi-locus sequence typing (MLST) scheme was used to subtype the isolates using BLASTn and 7 housekeeping genes (aroC, dnaN, hemD, hisD, purE, sucA, and thrA) [24].
Antimicrobial susceptibility testing (AST) of the S. Enteritidis
isolates was performed with the broth microdilution method (Lot: D-012XS, Xingbai
Biotechnology, Shanghai, China) and interpreted according to guidelines from the
2017 Clinical and Laboratory Standards Institute (CLSI) and the European
Committee on Antimicrobial Susceptibility Testing (EUCAST; https://eucast.org/).
A bacterial suspension with a precisely adjusted concentration of 0.5 McFarland
units was first prepared. The recommended protocol for introducing the control
sample and thoroughly amalgamating it with the bacterial solution was
meticulously adhered to. Subsequently, the resultant mixture was kept in an
incubation chamber at a constant temperature of 35 °C for 18 to 24
hours. This process was carefully conducted with the ultimate aim of determining
the minimum inhibitory concentration (MIC). Susceptibility to the following
antimicrobials was assessed:
Illumina reads were mapped to the reference genome using Bowtie 2 v2.2.8 (Johns
Hopkins University, Baltimore, MD, USA), SNPs were identified using Samtools v1.9
(Shanghai Institute of Life Sciences, Shanghai, China), and the data was combined
as described previously [25]. All high-quality SNPs (HQ snps) supported by
Statistical analysis was performed using SPSS software (Version 16.0, IBM,
Armonk, NY, USA). Comparisons of antibiotic resistance between clinical isolates
and chicken-origin isolates were performed by Chi-square/Fisher exact test for
categorical variables. p
All 83 isolates were identified as S. Enteritidis ST11 by MLST after WGS analysis (see Supplementary Table 1).
A total of 124 virulence genes were detected in the 83 Salmonella genomes. All 83 isolates were positive for 111 virulence genes. Seven known virulence factors showed different distributions in the VFDB matrix (rck, pefB, pefD, pefC, spvR, spvC, spvB), with a low carriage rate in chicken isolates (see Supplementary Table 1). These virulence genes are located on the virulence plasmid of S. Enteritidis.
Analysis of the antibiotic resistance phenotypes of S. Enteritidis
isolated from different sources revealed a minor level of resistance in the
clinical isolates. The multiple drug resistance rate of chicken-origin isolates
was higher than that of clinical isolates (p
| Antimicrobials | No. of resistant strains (%) | p value | ||
| Clinical isolates (n/48) | Chicken-origin isolates (n/35) | |||
| Ampicillin | 37 (77.1) | 32 (91.4) | 2.970 | 0.085 |
| Ceftazidime | 6 (12.5) | 5 (14.3) | 0.000 | 1.000 |
| Cefoxitin | 0 (0.0) | 1 (2.9) | - | 0.422 |
| Cefotaxime | 6 (12.5) | 6 (17.1) | 0.353 | 0.553 |
| Cefazolin | 27 (56.25) | 13 (37.1) | 2.960 | 0.085 |
| Ampicillin-sulbactam* | 32 (66.7) | 15 (42.9) | 4.672 | 0.031 |
| Imipenem | 0 (0.0) | 0 (0.0) | - | - |
| Tetracycline* | 9 (18.75) | 14 (40.0) | 4.563 | 0.033 |
| Nalidixic acid | 48 (100.0) | 35 (100.0) | - | - |
| Ciprofloxacin | 0 (0.0) | 3 (8.6) | 2.163 | 0.141 |
| Azithromycin | 0 (0.0) | 0 (0.0) | - | - |
| Chloramphenicol* | 0 (0.0) | 6 (17.1) | - | 0.004 |
| Gentamicin* | 0 (0.0) | 5 (14.3) | 4.991 | 0.025 |
| Pediatric compound sulfamethoxazole* | 2 (4.2) | 8 (22.9) | 5.025 | 0.025 |
| Multidrug resistance | 38 (79.2) | 32 (91.4) | 2.304 | 0.129 |
p values were calculated by chi-square analysis with SPSS version 16.0; * denotes a statistically significant difference.
A maximum-likelihood tree of all 83 S. Enteritidis strains was
constructed using SNPs (see Fig. 1). The phylogenic tree was broadly divided into
four subclades. Of note, the genomes of MDR and non-MDR strains were distributed
into different subclades. Subclade A strains were resistant to two groups of
antibiotics, except strain FC9132 which was resistant to 6 groups of antibiotics.
All strains of subclade B were resistant to two or three groups of antibiotics.
Some strains of subclade C were resistant to 5 or 6 groups of antibiotics, and
were resistant to
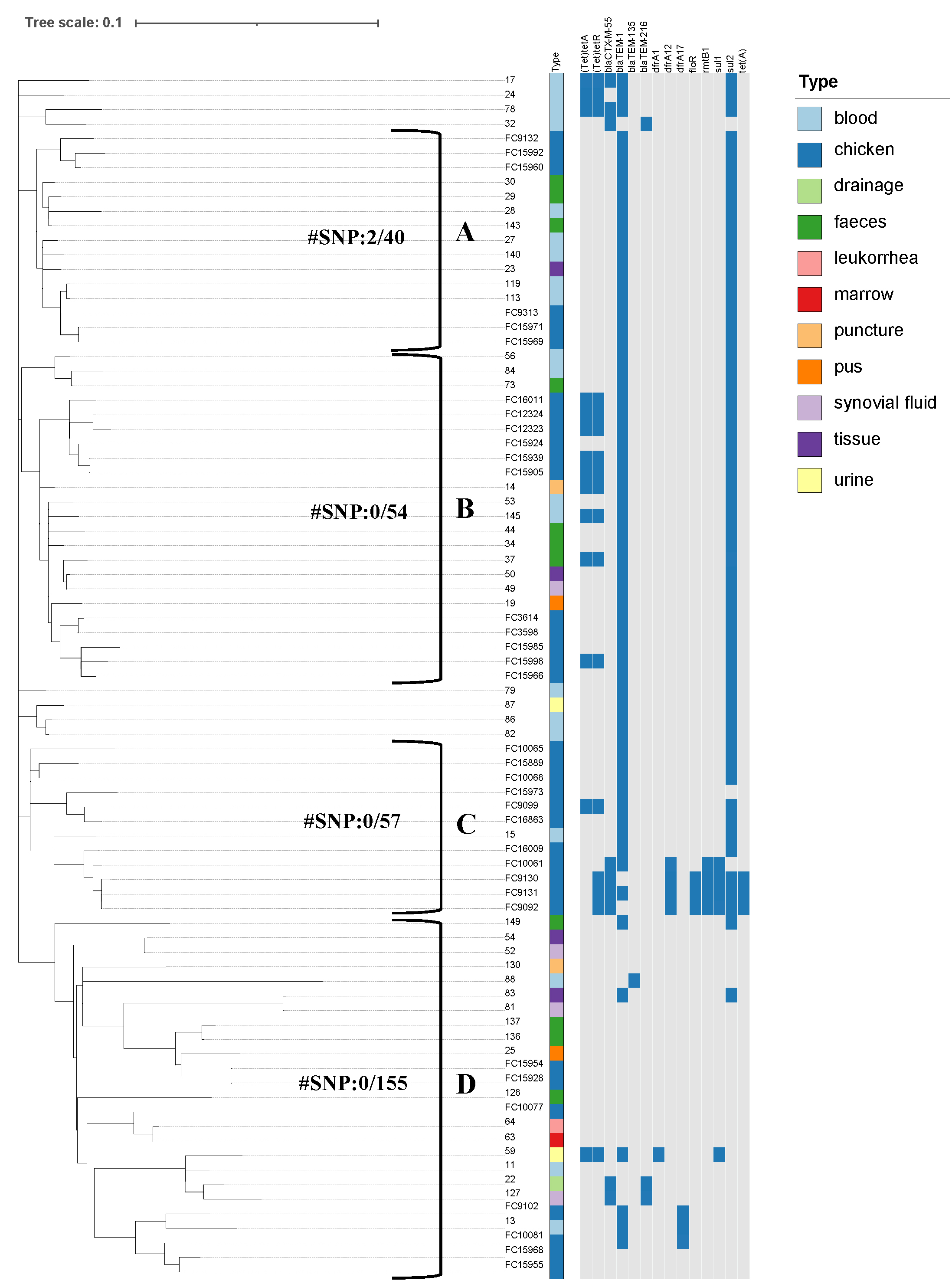 Fig. 1.
Fig. 1.Phylogenetic analysis showing antibiotic resistance patterns and the profiles of specific genetic loci in S. Enteritidis strains. Neighbor-joining tree based on whole-genome level single-copy genes in the 83 S. Enteritidis strains. The type indicates the different isolation source of strains, and the year indicates the year of isolation. The presence or absence of specific loci are denoted by blue and grey rectangles, respectively. The phylogenetic tree was divided into (A), (B), (C), and (D) subclades, with the adjacent values representing the minimum and maximum single nucleotide polymorphism (SNP) divergence between any two pairs of clades.
The 20 resistance genes associated with resistance tests were the
We next screened for mutations within the Quinolone resistance determination
region (QRDRs). All strains carried the gyrA mutation, with the mutation
types being D87Y (33/48 clinical isolates vs 28/35 chicken-origin
isolates), D87N (10/48 vs 3/35), and D87G (5/48 vs 4/35). No
significant differences in the mutation types were observed between the different
isolates (p
The WGS data was used to detect the presence and absence of transposons, plasmid replicons, and virulence genes in the 83 Salmonella genomes. Six types of plasmid replicons were detected: IncI1, col, IncN, IncX1, IncFIB, and IncFII. Of these, IncFIB (90.4%), IncFII (89.2%), and IncX1 (79.5%) were the most common. Only one strain (FC15968) had no plasmids, but was resistant to ampicillin and nalidixic acid. Three strains (FC15966, 17, and 78) that showed multiple drug resistance carried four types of plasmids and were resistant to 7, 7, and 3 types of antibiotics, respectively. Strains were divided into three clusters in the Inc_type matrix graph (see Fig. 2). In cluster A, the vast majority of strains were resistant only to NAL, and carried plasmids IncFIB and IncFII. In cluster C, most strains were resistant to two groups of antibiotics (with some up to 6 groups), and carried plasmids IncX1, IncFIB, and IncFII. Although most isolates in cluster B carried only the IncX1 plasmid, they still showed MDR.
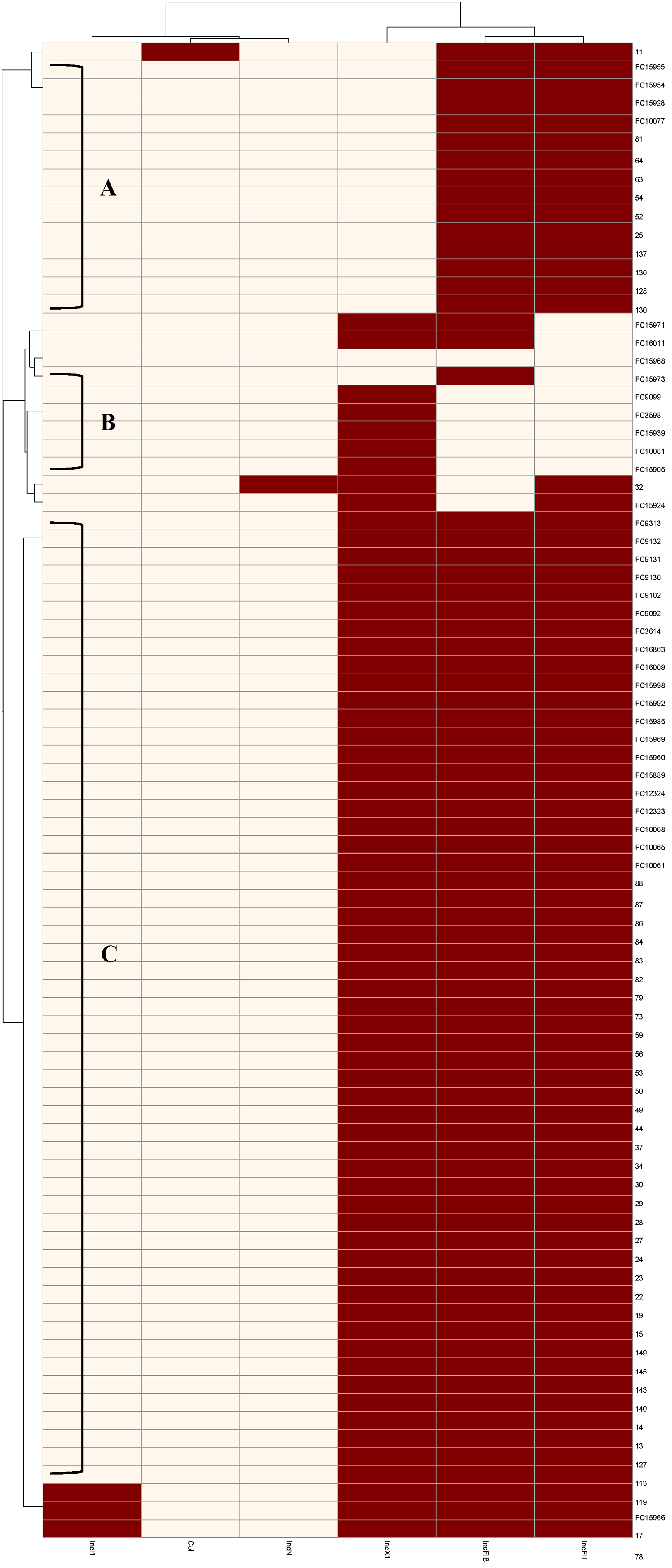 Fig. 2.
Fig. 2.Distribution of Inc types in different strains. The abscissa shows the Inc type and the ordinate shows the strain number. The presence or absence of Inc are denoted by red and beige rectangles, respectively.
We next analyzed the insertion sequence data for the 83 S. Enteritidis strains. A total of 562 insertion sequences were found, including ISEcp1, ISCro1, IS1R, ISSso4, and IS186B unique to aseptic clinical sites, and IS5075, ISVsa3, and ISCR1 unique to chicken. Most strains contained ISSaen1, IS1230B, IS285, IS26, ISSwi1 and IS1351. Isolates that were resistant only to nalidixic acid showed the insertion pattern ISSaen1-IS1230B-IS285-IS1351-IS1351. Some strains that were resistant to two or three types of drugs carried IS26 and ISSwi1, while all strains resistant to four types of drugs carried IS26 and ISSwi1 (except strain 59). FC9130 and FC9131 were resistant to 6 groups of drugs and carried the additional insertion sequence of IS50R-IS1294-IS5075-ISVsa3-ISCR1.
The only subtype of the bla
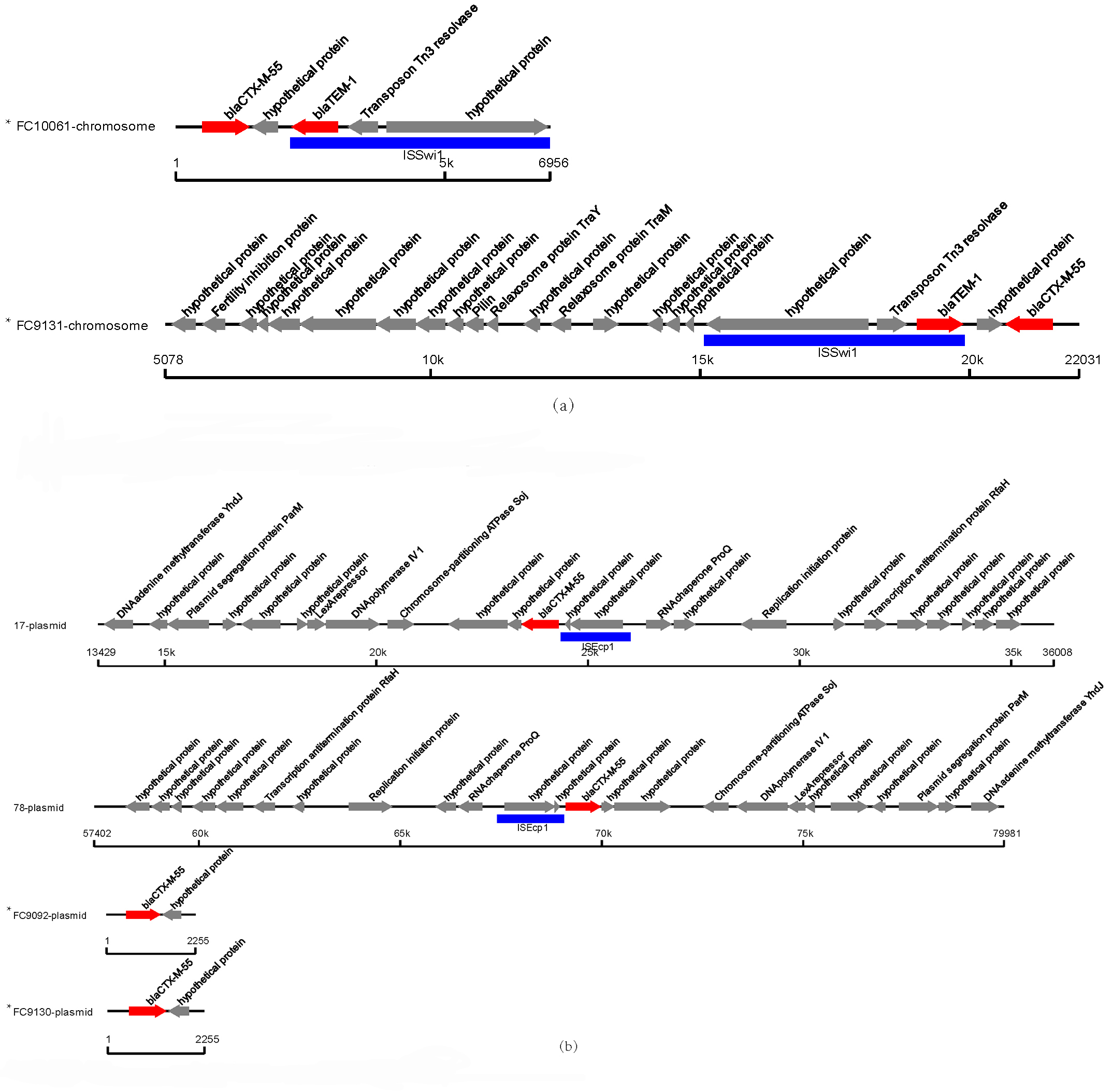 Fig. 3.
Fig. 3.Genetic environment of bla
The most common
A total of 8 isolates carried the drfA resistance gene, of which two
strains were clinical isolates (all located on plasmids) and 6 strains were
chicken-origin isolates (four strains located on plasmids and two strains located
on chromosomes, all belonging to the dfrA17 subtype) (see Fig. 4). The
two strains (FC10081 and FC9102) with dfrA17 located on the chromosome
had similar genetic contexts to the one strain (strain 13) located on the
plasmid. A total of 56 S. Enteritidis isolates had the sul2gene (29 clinical isolates and 27 chicken-origin isolates). The sul1
gene was found in one clinical isolate and in four chicken-origin isolates. All
sul1 and sul2 genes were located on plasmids, with no
significant difference in frequency between clinical and chicken-origin isolates
(p
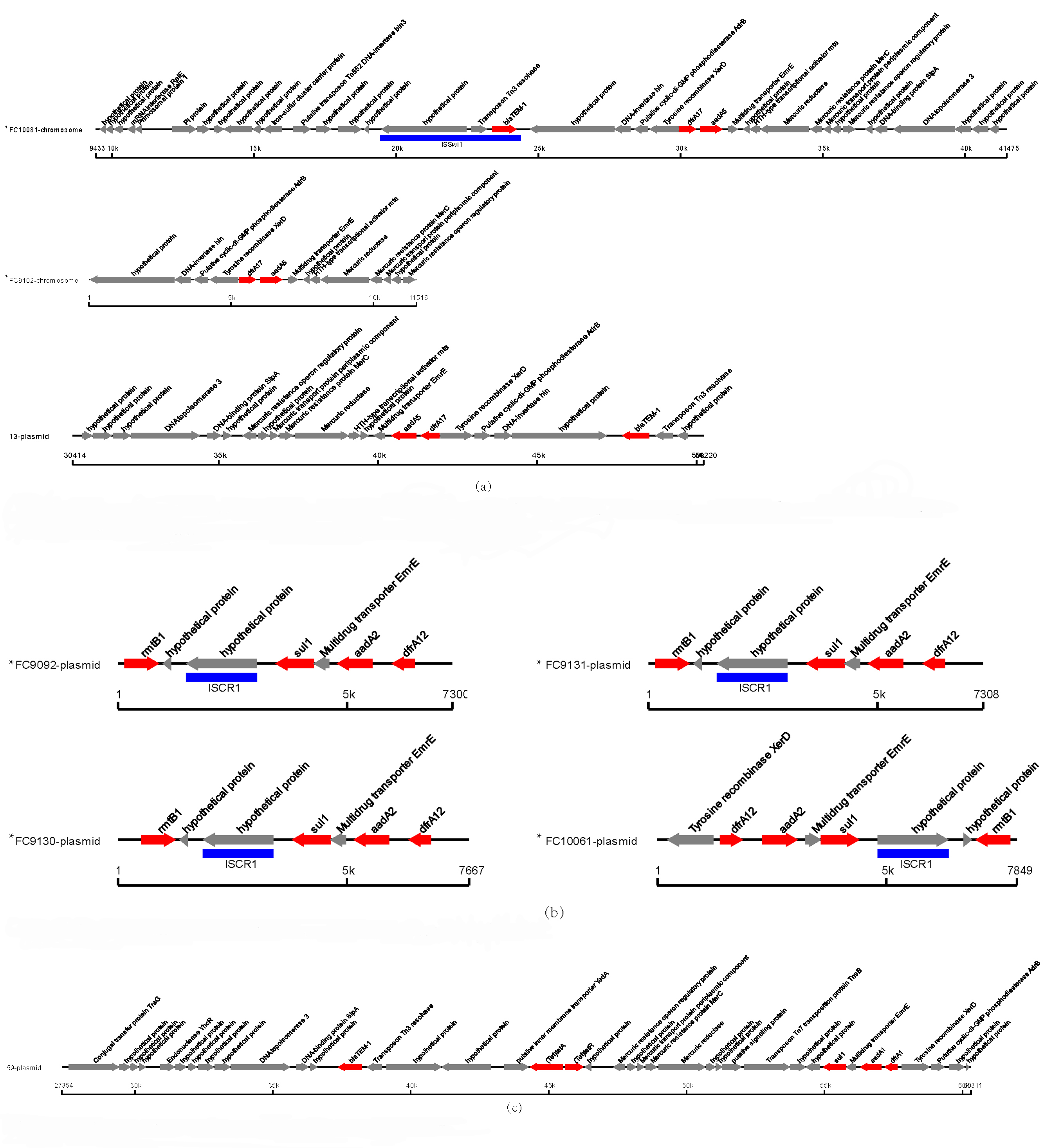 Fig. 4.
Fig. 4.Genetic environment of drfA in eight S. Enteritidis isolates (FC10081, FC9102, 13, FC9092, FC9130, FC9131, FC10061, and 59). (a) Comparison between dfrA17-positive strains. The dfrA17 of strains FC10081 and FC9102 were located on the chromosome. The dfrA17 of strain 13 was located on the plasmid. (b) The dfrA12 of strains were located on the plasmid. (c) The dfrA1 of strain was located on the plasmid.
Three S. Enteritidis isolates, all of chicken-origin, carried the floR gene on the plasmid, thereby conferring a higher resistance rate to chloramphenicol in the chicken-origin isolates compared to clinical isolates.
Seventeen strains carried tet(A) resistance genes, including 7 clinical
isolates and 10 chicken-origin isolates. In all cases the genes were located on
plasmids, with no significant difference in the frequency between the clinical
and chicken isolates (p
Several highly resistant chicken isolates were examined (strains FC15955, FC9092, FC9130, FC9131, FC9132, and FC10061). Most were found to carry special resistance genes such as floR, tet(A), sul1, and rmtB, and to carry more insertion sequences (e.g., IS1230B, IS285, ISSaen1) than other isolates.
Salmonellosis caused by S. Enteritidis is an important foodborne infectious disease [29]. The current study investigated 83 S. Enteritidis strains isolated from different sources. All S. Enteritidis strains were identified as ST11, consistent with the epidemiological characteristics of ST typing for S. Enteritidis [30, 31]. In addition, the results showed high genomic similarity between S. Enteritidis isolates from different sources and different regions, similar to the results of Davis et al. [32].
Differences in the virulence of pathogens can often be attributed to virulence factors [33]. To identify virulent S. Enteritidis isolates and to find genetic markers that distinguish invasive S. Enteritidis, we conducted a genome-wide association analysis of virulence factors using different sources of isolates as phenotypes. The three sources of isolates were aseptic sites in patients with extra-intestinal infections, feces from patients with diarrhea, and foodborne chicken-origin isolates. Screening of the VFDB database revealed a lower carriage rate for virulence genes from the Salmonella Virulence Plasmid in chicken-origin isolates compared to clinical isolates. This indicates the virulence plasmid plays an important role in the pathogenic process of S. Enteritidis.
A SNP-based phylogenetic tree describing the evolution of S. Enteritidis identified four distinct subclades. The differential resistance observed between subclade C and subclade D phenotypes implies the SNP differences may be because of differences in the resistance genes. We analyzed the resistance phenotypes of different isolates and found the MDR rate to be up to 83.1%, which was higher than a previous study [34]. Chicken isolates showed greater resistance to almost all antibiotics than clinical isolates, with a higher frequency of MDR. This indicates a broader drug resistance profile in the chicken-origin isolates. We are unable to speculate on possible differences in antibiotic use between the provinces from which the isolates were collected, and no clusters were visible in the phylogenetic tree according to the different provinces.
Analysis of the resistance genes indicated differences in the evolutionary characteristics between chicken-origin and clinical isolates. Several highly resistant chicken isolates (FC15955, FC9092, FC9130, FC9131, FC9132, and FC10061) were found to carry some resistance genes, such as floR, tet(A), sul1, and rmtB1. This may explain their greater resistance compared with other strains.
Resistance to quinolones is related to point mutations in QRDR, with the pattern
of point mutations in different genes being associated with different resistance
phenotypes and levels. Three mutations (D87Y, D87N, and D87G) were found in
gyrA, but none in parC and parE. Three strains
(FC15955, FC9131, and FC9132) found to be resistant to ciprofloxacin had the
D87G, D87Y, and D87Y mutations, respectively. The ciprofloxacin MIC values for
all other strains were
The CTX-M gene is associated with anti-cefotaxime levels. Only one CTX-M gene,
CTX-M-55, was detected in 9 isolates (strains FC9131, 127, 32, FC10061, FC9092,
78, FC9130, 22, and 17). No difference was observed in the prevalence of CTX-M-55
between clinical isolates and chicken-origin isolates. However,
bla
Lei et al. [41] analyzed the genetic relationship and antimicrobial resistance of S. Enteritidis strains isolated from Hebei province by WGS. Their isolates had a high resistance rate to NAL and AMP, and demonstrated the vertical transmission of S. Enteritidis from breeding chickens to commercial chickens. Li et al. [42] conducted a genomic analysis of dead poultry in Shandong province from 2008 to 2019. The authors found that isolated Salmonella strains showed greater resistance to ampicillin than to nalidixic acid. These findings are consistent with our results showing that chicken-origin isolates have high resistance to ampicillin and nalidixic acid, with even higher rates than those reported in earlier studies. Another study of resistance in samples from chicken meat products reported resistance to tetracycline only. The proportion of drug-resistant strains in the clinical isolates analyzed here was not particularly high compared to clinical isolates from other regions. A study conducted in 2013–2014 at the Hangzhou pediatric hospital reported a similar incidence of resistance to ampicillin as the present study [43]. Several studies have suggested that foodborne Salmonella enteritis may be the cause of clinical disease [36, 44]. Other studies have proposed that co-selection and transmission of ESBL and fluoroquinolone resistance in Salmonella occurs through the food chain, and that transmission of resistance genes is mediated by plasmids [45, 46]. A genomic surveillance study found that a pork isolate differed from a human isolate by only 10 SNPs, indicating that pork food was the likely source of human infection. Phylogenetic analysis suggests that foodborne Salmonella could be the source of clinical disease [47]. This Chinese study was based on enteritis caused by foodborne Salmonella and clinical pathogenic isolates analyzed in genome evolution, different sources isolates biggest differences in SNP number only about 155, and the drug-resistant genes may spread through the plasmid and insert sequences. We propose that foodborne MDR Salmonella may be transmitted to humans, and that antimicrobial resistance originating in chickens is concerning and its genomic evolution should be monitored.
Based on WGS data, this study compared virulence factors and drug resistance
between S. Enteritidis isolates of chicken-origin and clinical origin.
In contrast to clinical isolates, some chicken-origin S. Enteritidis
isolates did not carry the virulence plasmid that plays a role in pathogenesis.
Chicken-origin isolates were more resistant to drugs compared with clinical
isolates, with several strains being resistant to
The genome sequences in this study were deposited into the National Center for Biotechnology Information database under BioProject accession no. PRJNA918954.
JL: Conceptualization, Methodology, Data curation, Funding Acquisition, Writing - Review & Editing. YH: Conceptualization, Methodology, Formal Analysis, Visualization, Writing - Original draft preparation. LZ: Methodology, Data curation, Writing - Review & Editing. GC: Software, Validation, Writing - Review & Editing. JC: Methodology, Writing - Original draft preparation. QZ: Software, Validation, Writing - Review & Editing. LY: Methodology, Data curation, Writing - Review & Editing. SC: Conceptualization, Methodology, Data Curation, Writing - Reviewing and Editing. CW: Conceptualization, Methodology, Data Curation, Writing - Reviewing and Editing. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Oral informed consent was obtained for all strains collected from the People’s Liberation Army General Hospital between 2010 and 2017. The project had passed ethical review at the time of application, and when the experiment was carried out, it was approved by Beijing Chaoyang District Center for Disease Control and Prevention Ethics Committee (IRB Approval Number: CYCDPCIRB-20240312-1).
Not applicable.
Supported by the National Natural Science Foundation of China (82002206).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.