
- Academic Editor
Osteoarthritis (OA) is now considered as a multifaceted disease affecting
various articular tissues, including cartilage, bone, synovium, and surrounding
ligaments. The pathophysiology strongly implicates intricate chemical
communication, primarily through cytokines, leading to the production of
degradative enzymes in cartilage, inflammatory peptides in synovium, and
structural changes in bone, resulting in characteristic clinical features such as
joint deformities and loss of cartilage space seen on X-rays. Recent studies
highlight the previously underestimated role of subchondral bone in OA, revealing
its permeability to cytokines and raising questions about the influence of
abnormal perfusion on OA pathophysiology, suggesting a vascular component in the
disease’s etiology. In essence, alterations in bone perfusion, including reduced
venous outflow and intraosseous hypertension, play a crucial role in influencing
the physicochemical environment of subchondral bone, impacting osteoblast
cytokine expression and contributing to trabecular remodeling, changes in
chondrocyte phenotype, and ultimately cartilage matrix degeneration in OA.
Dynamic contrast (gadolinium) enhanced magnetic resonance imaging (DCE-MRI) was
used to quantify perfusion kinetics in normal and osteoarthritic subchondral
bone, demonstrating that decreased perfusion temporally precedes and spatially
correlates with cartilage lesions in both young Dunkin-Hartley (D-H) guinea pigs
and humans with osteoarthritis. Pharmacokinetic analysis of DCE-MRI generated
data reveals decreased tracer clearance and outflow obstruction in the medial
tibial plateau of osteoarthritic guinea pigs, coinciding with progressive
cartilage degradation, loss of Safranin O staining, and increased expression of
matrix metalloproteinases and interleukin-1. Positron emission tomographic (PET)
scanning using
Osteoarthritis (OA) is considered to be a disease of all articular tissues, involving bone, cartilage, synovium and the fibrous capsule and surrounding ligaments. The pathophysiology of OA involves prominently, crosstalk or, chemical communication by cytokines among these tissues, stimulating the production of degradative enzymes particularly in cartilage, inflammatory peptides particularly in synovium, and structural extracellular matrix proteins particularly in bone. These chemical communications culminate in the pathophysiology of OA – subchondral bone plate hypertrophy, trabecular osteoporosis, cartilage matrix degradation, synovial inflammation, and ligamentous-capsular fibrosis and contracture which, in turn, produces the structural hallmarks of OA, cartilage degradation and subchondral bone sclerosis (Fig. 1). Clinical characteristics include angulatory deformities of joints with loss of articular cartilage space and bone sclerosis on X-ray (Fig. 2).
 Fig. 1.
Fig. 1.Cartilage and Bone in Osteoarthritis. Subchondral plate hypertrophy, trabecular osteoporosis, and focal cartilage loss characteristic of osteoarthritis.
 Fig. 2.
Fig. 2.Asymmetric Joint Loss in Osteoarthritis. (Left) loss of cartilage and bone from the medial tibiofemoral compartment and a varus tibio-femoral angle on radiography. (Right) a clinical example of varus knees.
Chemical communication between synovium and cartilage has been known for some
years notably, by the discovery of interleukin 1, named at the time, catabolin
[1]. In a straightforward but elegant experiment, this study showed that a
non-enzymatic cytokine elaborated from synovium could stimulate cartilage
breakdown but only with live chondrocytes because of the stimulation of
chondrocytic enzymes. Communication between bone and cartilage, on the other
hand, was thought for many years not to occur because the subchondral bone plate
was considered impermeable. More recently, it has been shown in several studies,
that the subchondral plate is not impermeable but that bone in general, and the
plate in particular, are quite conductive to a variety of cytokines permitting
chemical communication between cartilage and bone [2, 3]. These observations make
imperative the understanding of the pathophysiology of subchondral bone in OA.
This narrative review describes alterations in the perfusion of OA bone and the
responses by osteoblasts to changing vascular conditions of their
microenvironment, thus strongly suggesting that OA is influenced by abnormal
perfusion and is a disease with a prominent vascular component in its
etiopathology. Osteoblasts respond to changes in their physicochemical
environment and, in particular, hypoxia induced by decreased perfusion, by
producing a wide range of cytokines that contribute to inflammation and matrix
remodeling in OA [4]. These cytokines can traverse through vascular and osseous
pathways, through the subchondral bone plate, and into the joint [2].
Consequently, the physicochemical microenvironment of the subchondral bone, and
in particular, environmental conditions induced by decreased perfusion, have
become of great interest in OA. This review presents dynamic measurements of
perfusion as a function of OA severity and indicates that venous outflow
obstruction is associated with increasing OA severity. It further presents
information that
The contemporary paradigm of the pathophysiology of OA is that a large number of signaling cytokines, degradative enzymes, and biomechanical stresses interact to produce the stereotypical picture of OA. Regarding bone, an important recognition has been that bone is a conductive medium for cytokine transit and that the subchondral bone plate is porous, facilitating communication between the cartilage and osseous compartments such that pathologies in one space can affect tissues in another (Table 1). These observations have stimulated interest in the physiochemistry of subchondral bone and the response of osteoblasts to alterations in their subchondral environment. Osteoblasts are very sensitive to physicochemical cues in the subchondral bone microenvironment and, in response to changes in perfusion including pressure, fluid flow, oxygen concentration, and pH, alter their production of cytokines in ways that contribute to the pathology of OA [5]. Osteoblasts subjected to oxygen concentrations of 35–40 mmHg, levels of hypoxia seen in OA, alter their synthesis of cytokines, signaling peptides, and growth factors, including vascular endothelial growth factor (VEGF), insulin-like growth factor-2 (IGF-II), transforming growth factor beta (TGFß1), and tissue inhibitor of metalloprotease (TIMP-1) that are associated with bone remodeling, and cartilage degeneration, the histopathologic characteristics of OA. Activation of cell signaling pathways in osteoblasts including inflammatory peptides, (cyclooxygenase (Cox2), prostaglandin E2, and nitric oxide (NO), transcription factors (c-Fos and the early growth response protein (Egr1))), as well as enzymes, notably, matrix metalloproteinase-1, 3 (MMP-1, 3), and 13 occurs in response to reductions in subchondral perfusion and elevated intraosseous pressure [5, 6]. OA osteoblasts also express elevated levels of cytokines and proteins that contribute to bone formation notably, type 1 collagen, osteocalcin, insulin-like growth factor-1 (IGF-1), and alkaline phosphatase, and that participate in remodeling of subchondral trabeculae and the subchondral bone plate. These are also responsible for the changes in bone structure observed in OA. OA osteoblasts also express elevated levels of cytokines that promote articular cartilage degeneration. OA osteoblasts produce a change in the chondrocyte phenotype that includes hypertrophy and matrix calcification [7]. Chondrocytes in co-culture with OA osteoblasts demonstrate reduced expression of cartilage type 2 collagen, parathyroid hormone related protein and receptor (PTHrP/PTH-R) and the SOX9 gene, and increase the expression the matrix-degrading enzymes, MMP-3 and 13 while down regulating aggrecan synthesis [8]. Matrix metalloproteinases catalyze the degeneration of cartilage matrix. Although they can be active in cartilage homeostasis, MMP-3, among others, is primarily seen in pathologic cartilage [9]. MMP-3 levels have been shown to be associated with, and to be an independent predictor for, cartilage matrix damage and may serve as a potential prognostic biomarker for cartilage injury in osteoarthritis patients [10].
| Subchondral Plate | Absence of a barrier between intra-articular and subchondral compartments: |
| - Allows chemical communication between cartilage and bone | |
| - Exposes articular cartilage to cytokines and enzymes from subchondral bone | |
| Subchondral Bone | Osteoblasts change their cytokine expression in response to their physicochemical microenvironment notably, hypoxia and in OA: |
| - Contribute to bone remodeling | |
| - Induce cartilage matrix breakdown |
In summary, bone perfusion has profound effects on the subchondral bone physicochemical microenvironment. Decreased perfusion and hypoxia, venous occlusion, or intraosseous hypertension, have profound influences on the cytokine expression profile of osteoblasts which can result in alterations in the chondrocyte phenotype and trabecular remodeling that can contribute to articular degeneration in OA [11]. As a consequence, it is important to understand the kinetics of subchondral bone perfusion that could contribute to the pathology of osteoblasts in OA.
Interest in the relationship of perfusion, especially venous stasis, to OA was
stimulated at St. Thomas’ and Guy’s Hospitals in the UK by Prof. Murray Brookes.
Work by Dr. Brookes and his team demonstrated that venous occlusion, or stasis,
had profound effects on bone and cartilage formation leading to OA. Brookes
employed a preclinical venous ligation model to demonstrate that decreased venous
outflow was associated with decreased subchondral pO
The D-H guinea pig exhibits the stereotypical cartilage lesions of human OA – loss of Safranin-O histochemical staining, articular fibrillation, and cartilage matrix clefts. Because OA develops spontaneously in the D-H guinea pigs and progresses according to a predictable time course, related to age of the animal, it is useful for the study of the pathogenesis of OA and potentially for the evaluation of disease-modifying agents [17, 18]. In this model, OA occurs spontaneously without the need for surgical or traumatic intervention. Its temporal and structural features have been well-described providing a physiological platform for the exploration of pathology of OA (Fig. 3) [19]. Early cartilage lesions may be apparent by 9 months of age with obvious cartilage lesions and subchondral sclerosis apparent by 12 months of age; by 16 to 18 months of age, subchondral sclerosis and cartilage degeneration are observable primarily on the medial tibial plateau.
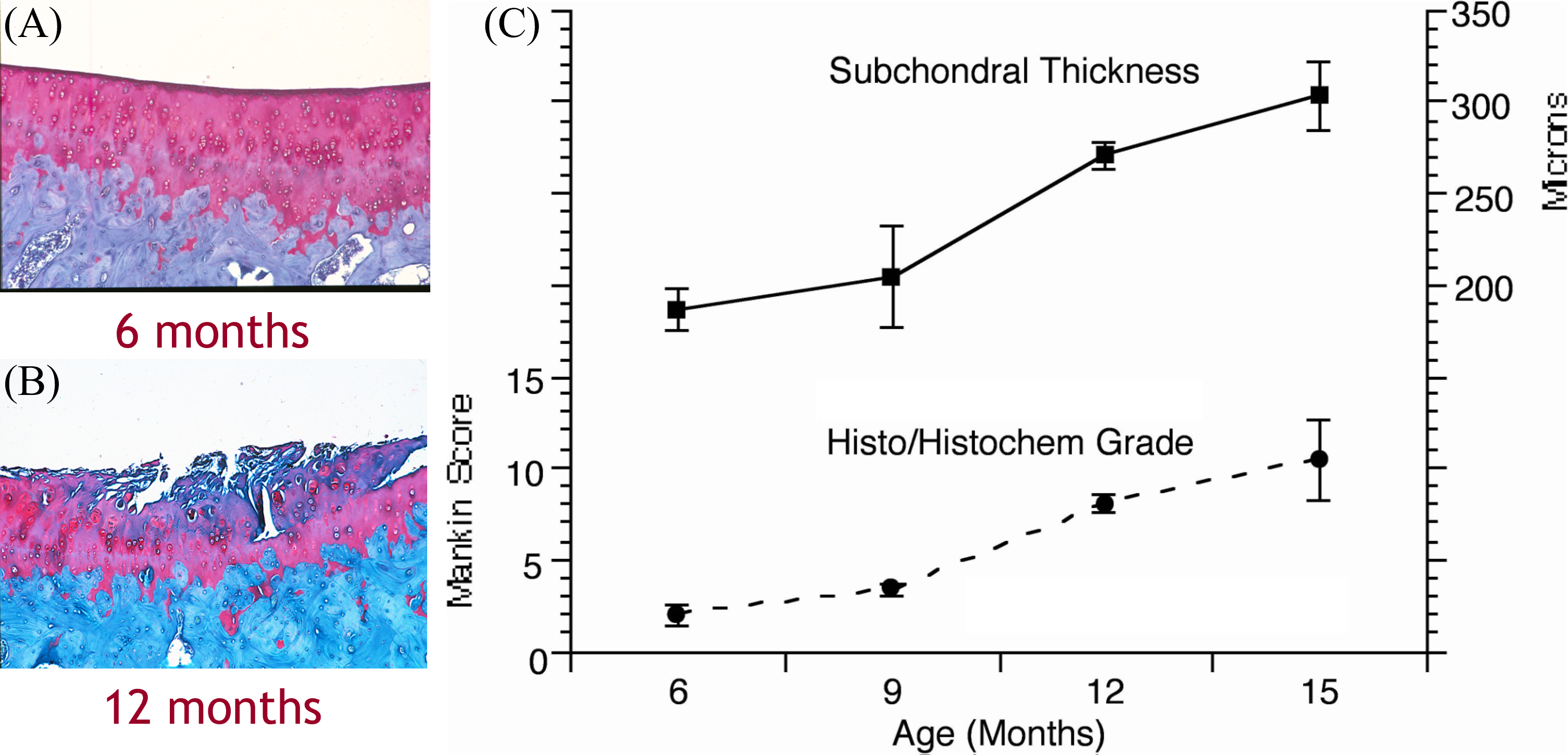 Fig. 3.
Fig. 3.Progression of Osteoarthritis in Dunkin-Hartley Guinea Pigs. (A) Normal cartilage with intact surface and Safranin O staining. (B) Surface fibrillation and loss of staining in osteoarthritis. (C) Characteristics of osteoarthritis beginning at 9 months of age and progressing substantially by 12 months of age.
Dynamic contrast enhanced magnetic resonance imaging (DCE-MRI) has been used to describe the relationship of subchondral bone perfusion to the development of OA in the D-H guinea pig model. The time course and spatial distribution of alterations in subchondral bone perfusion with developing OA have been characterized by comparing perfusion with the appearance cartilage lesions of developing OA [19]. In the D-H model, decreased perfusion temporally precedes the histopathologic characteristics of OA at 6–9 months of age (Fig. 4). Further, reduced perfusion is seen limited to the site of subsequent bone and cartilage lesions in the medial tibiofemoral compartment, leading to the conclusion that perfusion abnormalities both precede and anatomically localize at the location of eventual OA. By 9 months of age, when cartilage degeneration first appears but is, for the most part, superficial, delayed venous outflow is seen on DCE-MRI time-intensity curves, indicating that the primary circulatory event in OA pathophysiology is venous stasis [20]. These studies indicate that, in the D-H guinea pig OA model, venous stasis and associated compromised perfusion temporally precede and spatially localize with the eventual cartilage and bone extracellular matrix pathology. The observation of venous stasis has been supported by two other studies showing decreased elimination of contrast and venous stasis in both the rat femoral head and knee [21, 22].
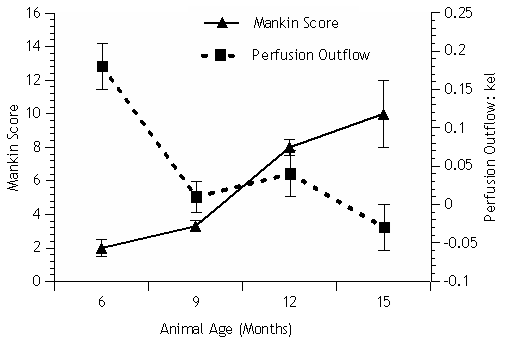 Fig. 4.
Fig. 4.Osteoarthritis developing between 9–12 months of age (Mankin
score) [20]. Decreasing venous perfusion outflow (k
Quantification of perfusion kinetics by DCE-MRI in normal subchondral bone has been compared to perfusion, cartilage degeneration, and bone remodeling in established OA in the D-H guinea pig [23]. Venous outflow obstruction, secondary intraosseous hypertension, reduced perfusion, and hypoxia occur in OA [13, 16]. It has been demonstrated, using DCE-MRI and pharmacokinetic modeling, that reduced perfusion spatially localizes with and temporally precedes eventual cartilage lesions in D-H guinea pigs [19]. Similar changes have also been shown to occur in human OA and avascular necrosis [24]. As described above, decreased perfusion of OA subchondral bone can alter the physicochemical environment of osteoblasts to which they respond by, (1) expressing altered cytokine patterns and remodeling the structural of both the subchondral trabeculae and bone plate and, (2) elaborating signaling cytokines which contribute to cartilage breakdown.
Several studies from our laboratory were conducted with a 3.0 Tesla GE HDx MRI Scanner. Animals were maintained in a custom device with a 7-loop, 3.2-cm-diameter inductively coupled solenoidal radiofrequency resonator. Data were analyzed using previously published in-house software based on a two-compartment model described by Brix [25]. The Brix model quantifies the kinetics of gadolinium exchange between the plasma and interstitial space [26]. Time intensity curves were created from contrast signal intensities within the region of interest (ROI). After DCE-MRI scanning, knees were excised, and tibias were decalcified. Coronal sections were cut at 5 µm thickness at the mid OA lesion or the central third of the tibia if no OA lesions were visible. Sections were stained with hematoxylin/Safranin O/fast green for histochemical analysis and graded by the criteria of Mankin [27]. OA lesions occurred primarily on the medial tibial plateau. The histological-histochemical scores described by Mankin were used to assess the severity of OA and ranged from 0–14 with higher scores indicating greater cartilage degradation. DCE-MRI time intensity curves characterized perfusion in the subchondral bone by decreased tracer clearance associated with increasing severity of OA (Fig. 5).
 Fig. 5.
Fig. 5.Time-intensity curves of contrast clearance in Dunkin Hartley guinea pig osteoarthritis based on age [19]. (A) At 6 months of age, before osteoarthritis, normal venous flow was indicated by an elimination constant of 0.06. (B) By 12 months of age, with established osteoarthritis, the elimination constant averaged –0.11, reflecting obstruction of venous outflow.
Three quantitative parameters were extracted from the compartmental fit of the
time intensity curve [23]. The amplitude, A, is related to the size of the
extracellular extravascular space; k
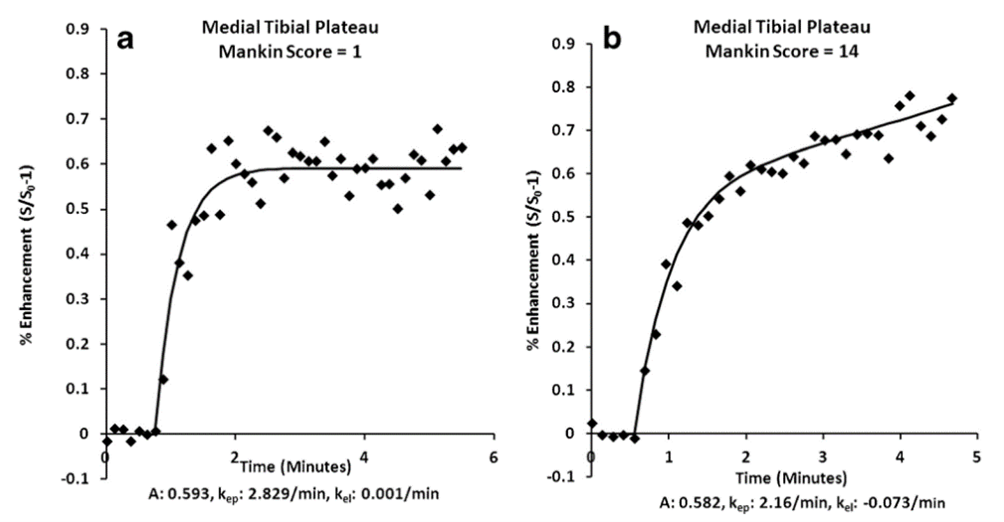 Fig. 6.
Fig. 6.Time-intensity curves based on severity of osteoarthritis [23]. (a) Normal perfusion demonstrated by a normal time-intensity curve in an animal with a Mankin score of 1 (minimal osteoarthritis). (b) Venous stasis shown by delayed clearance of contrast after 2 mins. of contrast infusion in an age-matched guinea pig with a Mankin score of 14 (severe osteoarthritis).
Coincident with decreasing tracer clearance and decreased perfusion, histochemistry revealed progressive loss of Safranin O staining, cartilage fibrillation, and eburnation in progressively severe OA [28]. Mean Mankin scores at 18 months of age were 11.7 +/–0.3. Immunohistochemistry revealed decreased aggrecan epitopes, and increased MMP-3 (stromelysin), MMP-13 (collagenase), and IL-1, consistent with cartilage breakdown. Another study, using 12-month-old Dunkin Hartley guinea pigs, described structural features of the developing OA and confirmed these observations [29]. Reduction in cartilage Safranin O staining, reduced cartilage thickness, and cartilage fibrillation, with relatively high mean Mankin scores of 10.9 +/–0.9 were observed in the medial tibial plateau cartilage.
PET with
In one study,
PET images displayed asymmetric isotope uptake as a function of the severity of
OA (Fig. 7). Time activity curves quantified uptake of
 Fig. 7.
Fig. 7.Positron emission tomographic (PET) scan using false colors to
delineate
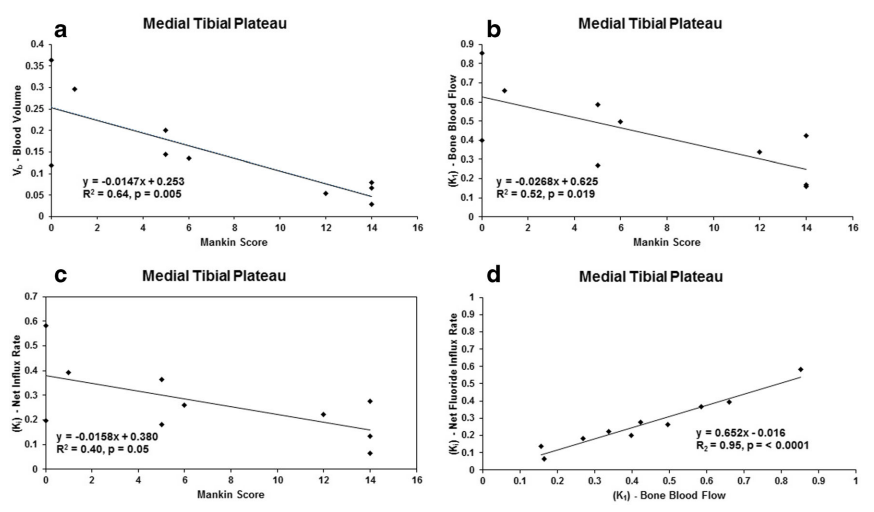 Fig. 8.
Fig. 8.The date derived from PET scanning confirm the observation that reduced blood
flow is associated with the severity of OA and extends that observation to
describe a relationship between decreased bone perfusion and abnormal bone
metabolism as shown by decreased
The relationships observed from DCE-MRI time intensity curves suggests an
enhanced and prolonged signal intensity and a reduction in the elimination
constant, k
The data obtained from PET scans support the findings that decreased subchondral
bone perfusion is associated with the severity of OA and demonstrates a
relationship between decreased perfusion and reduced osteoblast activity
(decreased
There are some limitations to this study. The research was conducted in a guinea pig preclinical model, although one with documented similarity to human OA. Models need to be chosen to display outcomes of interest and the D-H guinea pig model was temporally suitable to spontaneous OA and to be a source of OA tissue resembling that of humans. Blood flow was not measured directly but was inferred from well-established functional imaging models. The study focused on the relationship of perfusion in subchondral bone to OA. This is not to imply that other pathophysiologic events do not contribute to OA but to indicate that recent observations of the relationship of bone and cartilage is relatively underappreciated. Other articles in the special issue deal with complementary issues in OA pathology.
Imaging with DCE-MRI and
The emphasis of this review has been on the perfusion changes in subchondral
bone and the likely role they play in the pathogenesis of OA. Newer information
has delineated, by functional imaging altered perfusion of subchondral bone in
OA, the effects of perfusion alterations on the physicochemical microenvironment
of osteoblasts, the responses of osteoblasts to environmental changes, and the
porosity of bone creating a “cartilage-bone functional unit”. This report
delineates cartilage-bone interactions, stimulated by perfusion
alterations—important, but not exclusive, pathophysiologic contributions to OA.
It is important to note that crosstalk takes place between synovium and cartilage
has been observed for many years. In fact, as pointed out in the Introduction,
the chemical communication between synovium and cartilage was the first to be
identified in the context of OA pathology. In ensuing years, many other
inter-relationships between synovial and cartilage pathology have been
identified. Synoviocyte hyperplasia, hypertrophy, and fibrosis have been
accompanied by secretion of pro-inflammatory cytokines notably, IL-1and 6,
TNF-
This review has concentrated on the demonstration of perfusion alterations and osteoblastic metabolic consequences in a preclinical model of OA. It is to be noted that systemic vascular comorbidities coexist with knee OA in humans. Patients with osteoarthritis (OA) exhibit higher than expected prevalences of cardiovascular comorbidities including ischemic coronary disease, cerebrovascular and peripheral arterial disease, and venous thromboembolism [45, 46, 47]. Patients with OA are generally considered to be at higher risk of death from cardiovascular disease than individuals in the general population. The coincidence of obesity, hypercoagulation, type 2 diabetes, and inflammation in both OA and cardiovascular diseases has suggested shared similarities in their pathophysiology and phenotypes, possibly associated with the metabolic syndrome [48]. Structural and physiologic abnormalities have been demonstrated in retinal arterioles, and carotid and popliteal arteries in patients with generalized and hand OA [49, 50]. However, convincing evidence of arterial pathology is still elusive for both sexes and in hip and knee OA [50, 51]. A systematic review has delineated some of the similarities and uncertainties between Atherosclerotic Peripheral Vascular Disease (ASPVD) and OA [45]. However, the association between vascular pathology and knee OA has remained inconclusive because some studies reported a positive association between structural arterial pathology and OA while other studies have found no association between arterial pathology and knee OA [45]. The association between ASPVD and OA may be stronger in systemic hypertension. Knee OA is more prevalent among hypertensive individuals compared with normotensive individuals. A number of systematic reviews have indicated a closer relationship between hypertension and structural damage in knee OA than with OA pain [52]. Mechanistically, hypertension is thought to increase intraosseous pressure and contribute to hypoxia in the subchondral bone, altering osteoblast expression of both signaling and structural cytokines. This, in turn, contributes to bone remodeling and cartilage matrix breakdown [52]. Other studies have supported the observation that systemic hypertension is associated with OA, and one has suggested that antihypertensive medications could reduce the incidence of OA in hypertensive individuals [53]. The pathophysiological coincidences between ASPVD and OA are explored in more detail in another paper in this special issue.
IGF-1, insulin-like growth factor-1; VEGF, vascular endothelial growth factor; PTHrP, parathyroid hormone related protein; PTH-R, parathyroid hormone receptor; MMP, matrix metalloproteinase.
Conceptualization, RKA. Investigation, JO and RKA. Writing—original draft, JO and RKA. Writing—review and editing, JO, JPD, and RKA. Original reference work, JPD and RKA. Project administration, JO. Funding acquisition, RKA. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by a grant from the Miriam Hospital.
The authors declare no conflict of interest. Given the role as Guest Editor, Roy K. Aaron had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Emerito Carlos Rodriguez-Merchan.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.