
- Academic Editor
Background: The Mammalian Target of Rapamycin (mTOR) signaling pathway regulates protein phosphorylation and exerts control over major cellular processes. mTOR is activated by the small G-protein Ras Homolog Enriched in Brain (Rheb), which is encoded by the Rheb1 and Rheb-like-1 (RhebL1) genes. There is currently a paucity of information on the role of RhebL1, and specifically its involvement in viral infection. In the present study we investigated the role of RhebL1 during human influenza A/NWS/33 (NWS/33) (H1N1) virus infection of rhesus monkey-kidney (LLC-MK2) cells and human type II alveolar epithelial (A549) cells. Methods: To assess the efficiency of NWS/33 virus replication, the expression of viral nucleoprotein was examined by indirect immunofluorescence (IIF) and the viral yield by fifty percent tissue culture infectious dose assay. An RNA-mediated RNA interference approach was used to investigate the role of RhebL1 during NWS/33 infection. RhebL1 expression was evaluated by IIF, Western blotting, and enzyme-linked immunosorbent assays. A two-tailed Student’s t-test was applied to evaluate differences between groups. Results: RhebL1 was differentially expressed in the cell models used in this study. Silencing of the RhebL1 gene led to increased NWS/33 virus infection in A549 cells, but not in LLC-MK2 cells. Moreover, the expression of hyperphosphorylated cytokeratin 8, a marker of NWS/33 virus infection efficiency, increased in A549 cells depleted of RhebL1 but remained almost unchanged in LLC-MK2 cells. Conclusions: These are the first results showing involvement of the endogenous RhebL1 protein during viral infection. Our data suggests that RhebL1 exerts a host cell-dependent modulatory role during influenza virus infection. RhebL1 appears to be a restrictive factor against NWS/33 virus replication in A549 cells, but not in LLC-MK2.
Successful virus replication relies on specific strategies to gain control of key host cell signaling pathways. The Mammalian Target of Rapamycin (mTOR) is a highly conserved signaling pathway that plays a central role in consolidating the cellular physiology of all eukaryotes. It achieves this by controlling major processes such as protein synthesis, cellular growth/proliferation, and apoptosis [1, 2, 3, 4]. Hyperactivated mTOR signaling has been associated with cancer, diabetes, and the aging process [5, 6, 7].
mTOR constitutes the catalytic subunit of two distinct complexes known as mTOR complex 1 (mTORC1) and mTORC2. These have different substrates and functions [7]. Specifically, mTORC1 controls the balance between anabolism and catabolism, while mTORC2 governs cytoskeletal behavior and activates various survival and proliferation pathways [3]. Both complexes integrate upstream environmental information to gate their activation. mTORC1 is activated by the small G-protein Ras Homolog Enriched in Brain (Rheb) in its GTP-bound state, which is promoted by growth factors [8]. The Ras Homolog Enriched in Brain 1 (Rheb1) and Rheb-like-1 (RhebL1) genes are encoded by Rheb [9]. Although the role of Rheb1 has been widely established [8, 10, 11], little is known regarding the involvement of RhebL1 [12]. Moreover, most previous studies have investigated the transient over-expression of RhebL1, rather than the endogenous protein [13].
The mTOR pathway plays an important role in viral replication by regulating apoptosis, cell survival, and both transcription and translation mechanisms [14, 15]. Specifically, the 1918 pandemic strain of influenza A virus requires mTORC1 activity during the early replication phases [16]. Moreover, influenza A virus modulates the levels of mTOR RNA and protein [17].
Kim et al. [18] studied the involvement of RhebL1 in keratin 8 phosphorylation and reorganization in a human type II alveolar epithelial cell line (A549). In this regard, we previously reported that influenza A/NWS/33 (NWS/33) virus induces keratin 8 hyperphosphorylation in A549 cells, thereby enhancing its replicative efficiency [19].
The rhesus monkey-kidney (LLC-MK2) (simian) and A549 (human) mammalian cell models were previously reported to show different levels of permissiveness to influenza A virus infection [19, 20]. The aim of the present study was therefore to compare the involvement of RhebL1 protein during NWS/33 virus infection in these two cell models. To achieve this, we evaluated the effect of small interfering RNA (siRNA)-mediated silencing of the RhebL1 gene on both viral replication efficiency and on the phosphorylation of keratin 8.
We found that RhebL1 was expressed at a higher level in A549 cells than in LLC-MK2 cells and has a host cell-dependent modulatory role in NWS/33 replication. More specifically, RhebL1 acts as a restriction factor against NWS/33 virus replication and keratin 8 hyperphosphorylation in A549 cells, but did not to appear to have an active modulatory role in LLC-MK2 cells.
A549 (TCL 101), LLC-MK2 (BS CL 57), and Madin-Darby canine-kidney (MDCK, BS CL
64) cells were obtained from the Lombardy and Emilia Romagna Experimental
Zootechnic Institute (IZSLER, Brescia, Italy). Cells were cultured in either
Ham’s F-12 Nutrient Mixture (A549) or Earle’s Modified Eagle’s Medium (LLC-MK2
and MDCK) containing 2 mM L-glutamine, 10% fetal bovine serum, and antibiotics
(100 U/mL penicillin and 100 µg/mL streptomycin). All cell lines were
validated at IZSLER by Short Tandem Repeat-DNA typing and tested negative for
mycoplasma, as per international guidelines. Cells were cultured in a humidified
incubator at 37 °C and 5% CO
Human influenza A/NWS/33 virus (H1N1; ATCC VR 219) was propagated as described previously [21]. In brief, LLC-MK2 and A549 cells were grown to confluence in shell vials or 6-well plates and then infected at a multiplicity of infection (MOI) of 0.1 and one plaque-forming unit (PFU)/cell, respectively. After adsorption for 75 min at 4 °C, the viral inoculum was removed and the cells washed twice with serum-free medium before incubation for the time indicated.
Monolayers of LLC-MK2 and A549 cells were fixed and permeabilized in methanol
for 5 min at –20 °C. The cells were then washed with phosphate-buffered
saline (PBS, pH 7.4; 7 mM Na
For IIF assays, mouse monoclonal anti-influenza A virus nucleoprotein (NP) (1:30, BioMérieux), goat polyclonal anti-RhebL1 (1:15; Santa Cruz Biotechnology, Bergheimer, Germany), and rabbit anti-phosphorylated keratin 8 on serine 431 (1:100, ThermoFisher Scientific, Waltham, MA, USA) antibodies were used as described previously [19]. These antibodies were in turn detected by Alexa Fluor 568 goat anti-mouse IgG (1:500; Molecular Probes, Eugene, OR, USA), Alexa Fluor 467 donkey anti-goat IgG (1:500; Molecular Probes), and fluorescein isothiocyanate-conjugated donkey anti-rabbit IgG (1:70; Li StarFish, Milan, Italy) antibodies, respectively.
For each cell monolayer, 10 arbitrarily selected fields were analyzed and viral
NP-positive cells were expressed as the mean percentage value of the total cell
number per field, as estimated by chromatin staining with
4
WB assays were performed as previously described [22] on cell lysates collected from two replicate wells for each experimental condition. Mouse monoclonal anti-beta-actin IgG (1:400; Santa Cruz Biotechnology) and rabbit polyclonal anti-Rheb2-N-terminal (1:300; Prodotti Gianni, Milan, Italy) antibodies were used for the WB assays. Bound antibodies were detected by anti-mouse (1:5000; Sigma-Aldrich) and anti-rabbit (1:5000; Sigma-Aldrich) IgG alkaline phosphatase-conjugated antibodies.
To obtain transient knockdown of the RhebL1 gene, we used siRNA
(5
The viral yields in culture supernatants from MDCK cells were assessed as previously reported [23].
To quantify RhebL1 protein, the human RHEBL1 (GTPase RhebL1) ELISA kit was employed according to the manufacturer’s guidelines (Fine Biotech Co., Wuhan, Hubei, China). First, a standard curve was created using a standard control and starting at a protein concentration of 1000 pg/mL, followed by 2-fold serial dilutions from 1/2 to 1/64. The RhebL1 concentration in samples was calculated based on the standard curve. Briefly, 100 µL of sample (or prepared standard points) was added to each dedicated well of a 96-well plate. This was sealed and incubated for 90 minutes at 37 °C, after which the plate was washed twice and 100 µL of biotin-labeled antibody working solution was added to each well. The plate was then sealed and incubated again for 60 minutes at 37 °C. After three washes, 100 µL of horseradish peroxidase-streptavidin conjugate working solution was added to each well before sealing the plate and incubating for 30 minutes at 37 °C. In the last step, the plate was washed five times and 90 µL of tetramethylbenzidine substrate solution was then added to each well. The plate was once again sealed and incubated for 15 minutes at 37 °C. Finally, 50 µL of stop solution was added to each well and the optical density (OD) absorbance at 450 nm was immediately read.
Regarding the OD calculation, the relative OD absorbance at 450 nm was obtained with the following formula:
relative OD
The standard curve was plotted as the relative OD
GraphPad Prism software (Version 10.0.0 (153), GraphPad Software LLC, Boston, MA, USA) was employed for the statistical analysis. A two-tailed
Student’s t-test was used to evaluate differences between cells under
study. p-values
The expression and cellular distribution of endogenous RhebL1 protein was first examined with the IIF assay in LLC-MK2 and A549 cells (Fig. 1A,B). RhebL1 expression was lower in uninfected LLC-MK2 cells than uninfected A549 cells. Furthermore, RhebL1 showed diffuse staining with prevalent accumulation in the perinuclear region (see insets in Fig. 1A,B).
 Fig. 1.
Fig. 1.Analysis of Ras Homolog Enriched in Brain Like-1 (RhebL1)
expression in uninfected and human influenza A/NWS/33 (NWS/33)-infected rhesus
monkey-kidney (LLC-MK2) and A549 cells. RhebL1 protein expression in uninfected
LLC-MK2 cells (A) and A549 cells (B) was investigated using indirect
immunofluorescence (IIF) assays. Images were recorded with a conventional
fluorescence microscope. Scale bar = 20 µm. (A,B) The insets shown
at the bottom of each figure represent the higher magnification (2000
ELISA was used to determine the concentration of RhebL1 in cellular homogenates of either uninfected or NWS/33-infected (MOI = 1 PFU/cell, 24 h) LLC-MK2 and A549 cells (Fig. 1C). The results showed much higher expression of RhebL1 in uninfected A549 cells than in uninfected LLC-MK2 cells. NWS/33 virus infection did not alter the RhebL1 concentration in LLC-MK2 cells, but caused a significant decrease in A549 cells.
Next, we used an RNA-mediated RNA interference approach to study the effect of RhebL1 depletion on NWS/33 virus infection in LLC-MK2 and A549 cells, as described in the Methods section. The effectiveness of RhebL1 depletion in both models was first assessed by WB (Fig. 2A).
 Fig. 2.
Fig. 2.RhebL1 protein restricts productive NWS/33 virus infection in
A549 cells. (A–C) LLC-MK2 and A549 cells were treated for 48 h with either
control siRNA or RNA-mediated interference against RhebL1. (A) RhebL1 expression
in LLC-MK2 and A549 cells was evaluated by Western blotting (WB), with beta-actin
used as the protein loading control. (B) Histogram showing the percentage of
viral NP-positive LLC-MK2 and A549 cells following treatment for 48 h with either
control siRNA or RhebL1 siRNA, as evaluated by IIF after infection with NWS/33
virus (MOI = 0.1 PFU/cell, 24 h). (C) The viral yields in the supernatants of
LLC-MK2 and A549 cells were evaluated by the fifty percent tissue culture
infectious dose (TCID
Subsequently, the control cells and cells depleted of RhebL1 were infected with
NWS/33 virus (MOI = 0.1 PFU/cell, 24 h) prior to the IIF assay. Compared to
control cells, the percentage of viral NP-positive cells increased significantly
in A549 cells depleted of RhebL1, as shown in Fig. 2B. Conversely, depletion of
RhebL1 in LLC-MK2 cells had no effect, or only a slightly negative effect on
NWS/33 virus infection. Using the TCID
We previously reported that NWS/33 virus infection can stimulate hyperphosphorylation of keratin 8 on serine 431, thereby promoting virus replication in A549 cells but not in LLC-MK2 cells [19]. In view of the involvement of RhebL1 in keratin 8 phosphorylation in A549 cells [18], we next evaluated the effect of RhebL1 gene silencing on the expression of phosphorylated keratin 8 in both cell models.
To this end, control cells and cells depleted of RhebL1 were examined by IIF (Fig. 3A–D). No apparent changes in the expression of phosphorylated keratin 8 were observed in LLC-MK2 cells (see Fig. 3B vs. 3A). In contrast, increased expression and perinuclear accumulation of phosphorylated keratin 8 were observed in A549 cell monolayers (see arrows in Fig. 3D).
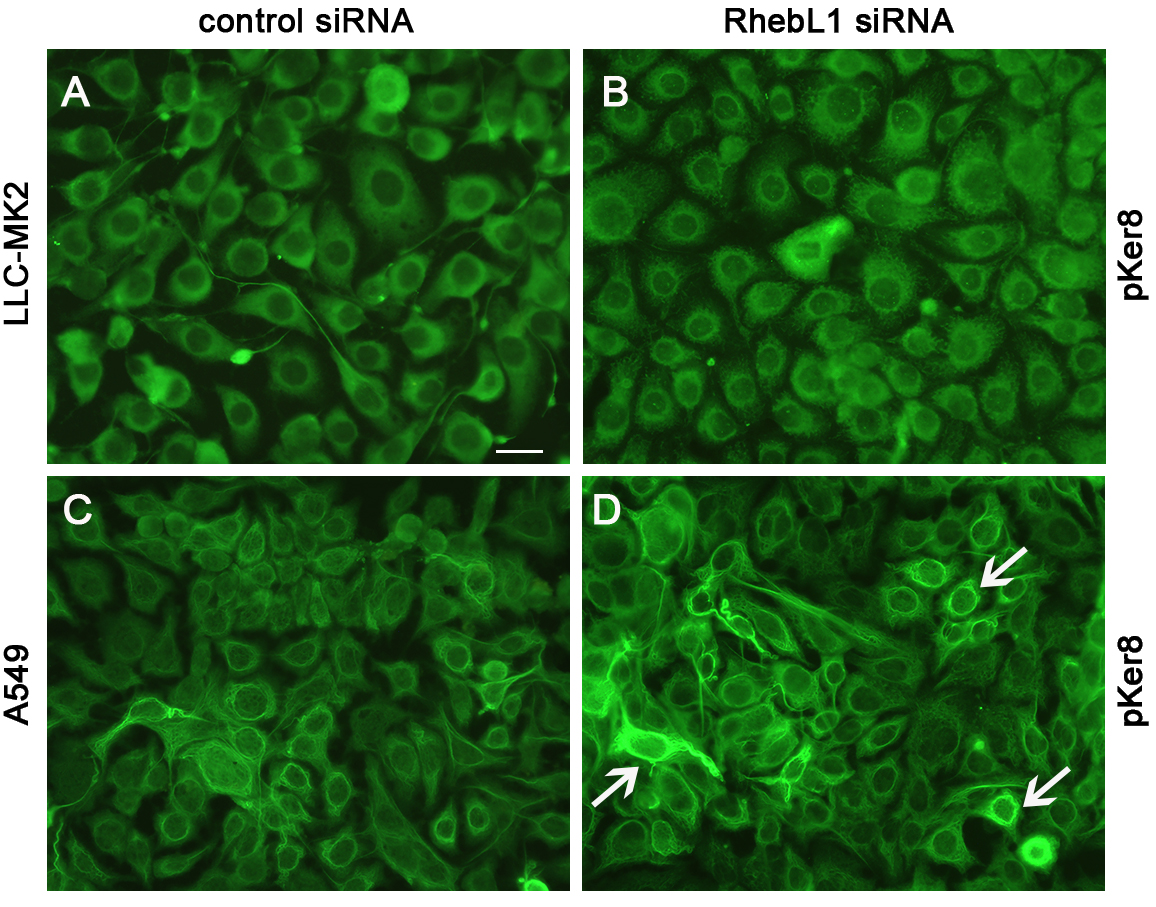 Fig. 3.
Fig. 3.RhebL1 protein restricts the expression of phosphorylated keratin 8 in A549 cells. LLC-MK2 and A549 cells were treated for 48 h with control siRNA (A,C) or with RNA-mediated interference (B,D). The expression of phosphorylated keratin 8 in LLC-MK2 cells (A,B) and in A549 cells (C,D) was then visualized by IIF. Images were recorded using a conventional fluorescence microscope. Scale bar = 20 µm.
Viruses are master manipulators of cell functions and can hijack key signaling pathways to modulate cell survival and ensure their replication. Since the mTOR signaling pathway is located at the crossroads of several fundamental cellular pathways, many viruses have developed specific mechanisms that target this major biological switch, thereby inducing metabolic reprogramming [24, 25].
The focus of the present study was on RhebL1, one of the modulators of the mTOR pathway, as to our knowledge there is no data regarding the involvement of this protein in viral infection. Several lines of evidence have implicated the mTOR pathway in viral infection. The influenza A virus is thought to induce autophagy by overwhelming the mTOR pathway, thereby leading to its successful replication [26, 27, 28]. Furthermore, the respiratory syncytial virus induces autophagy and suppresses the mTOR pathway to evade the host’s immune defenses, thus favoring its survival within the host [29]. Arunachalam et al. [30] also reported the mTOR pathway was downregulated in the plasmacytoid cells of subjects with Coronavirus disease 2019 (COVID-19). Conversely, Appelberg et al. [31] showed that replication of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) increased mTOR signaling, thereby ensuring the translation of viral proteins and allowing viral assembly [32].
The aim of this study was to evaluate the involvement of the RhebL1 protein, an activator of the mTOR pathway, during NWS/33 virus infection in two mammalian cell models with different levels of permissivity [19, 20].
RhebL1 is highly expressed in cells of the nervous system [33] and in other tissues [34], suggesting it has essential regulatory roles in a variety of cell types. The present study found a much higher level of RhebL1 expression in human A549 cells than in monkey LLC-MK2 cells. Given this differential expression, it is conceivable that RhebL1 performs different types of regulatory functions in these two cell models.
Specifically, RhebL1 has been shown to interact with molecules that activate signal transduction pathways for different protein kinases, including the phosphorylation of keratin [13, 18, 35, 36, 37, 38]. Similar to the effect of chemical activators of phosphorylation [19], RhebL1 gene silencing was shown to significantly increase the replication cycle of NWS/33 virus and the phosphorylation of keratin 8 in A549 cells, contrary to what is observed in LLC-MK2 cells.
Keratin 8 phosphorylation plays a significant role in regulating keratin filament organization, associations with binding proteins, and modulation of the cell cycle. Previous studies have emphasized the role of phosphorylation in viral pathogenesis. In this regard, Padilla-Mendoza et al. [39] focused on the expression of phosphorylated proteins, including keratin 8, and their association with the progression of cervical lesions caused by papillomavirus. Toivola et al. [40] demonstrated that site-specific keratin 8 phosphorylation is a marker of the progression of liver diseases caused by hepatitis C virus. We previously reported that increased keratin 8 phosphorylation on serine 431 favored virus replication during influenza A virus infection of A549 cells [19].
RhebL1 was recently found to induce robust phosphorylation of interferon regulatory factor 3, thereby acting as a trigger of innate immunity [41]. In this regard, our data indicates that RhebL1 has a negative regulatory role on NWS/33 virus infection in permissive A549 cells. Accordingly, we observed a significant decrease in virus-induced expression of RhebL1 at 24 h after NWS/33 virus infection in A549 cells, but not in LLC-MK2 cells. Moreover, RhebL1 gene silencing stimulated NWS/33 virus replication in A549 cells, but not in LLC-MK2 cells. Hence, we speculate that downregulation of RhebL1 in A549 cells might help the NWS/33 virus to decrease the activity of the host biosynthetic machinery, while favoring viral transcription and protein synthesis. This concurs with previous data showing that mTOR suppression during PR8 influenza virus infection increased viral transcription [42]. Ranadheera et al. [16] also observed downregulation of the mTOR pathway at early time points during influenza H1N1 (1918 strain) virus infection. However, this reverted to a steady state level at later post-infection time points, allowing the production of viral progeny.
With regard to the modulatory effects of RhebL1 and the possible impacts on virus infection, Yuan et al. [34] also reported a relationship with NF-kappa B-mediated gene transcription. Downregulation of RhebL1 induced by either viral infection or gene silencing could repress cellular antiviral responses triggered by NF-kappa B, as previously observed during influenza virus infection [43, 44].
Understanding the effects of host cell metabolic status on virus replication helps to identify mediators of viral tropism and to explain why some cells are more prone to virus infection than others. This is the first report on the host cell-dependent regulatory role of RhebL1 protein during influenza A virus infection. Our study has revealed new information on the complex metabolic landscape of cells infected with the influenza virus. This is worthy of further investigation with a view to developing broad-range antiviral therapies.
A549, human type II alveolar epithelial; ACRYL, acrylamide; BSA, bovine serum
albumin; DAPI, 4
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
FDC designed the research study. MB and FDC performed the research. MB and FDC analyzed the data. MB and FDC wrote the manuscript. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by grants allocated to Flora De Conto by the Italian Ministry of Education, University and Research (MIUR) (“Fondo per il finanziamento delle attività di base di ricerca (FFABR) – 2017”). The founders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.