
- Academic Editor
†These authors contributed equally.
Most pancreatic cancers are pancreatic ductal adenocarcinomas. This is an extremely lethal disease with poor prognosis and almost no treatment choices. Considering the profound role of the pancreas in the human body, malfunction of this organ can significantly affect quality of life. Although multiple metabolic pathways are altered in cancer cells, certain metabolic gene signatures may be critical for immunotherapy. The reprogrammed metabolism of glucose, amino acids, and lipids can nourish the tumor microenvironment (TME). Previous studies have also shown that reprogrammed metabolism influences immune responses. Tumor-infiltrating immune cells in the TME can adapt their metabolism to blunt the immune system, leading to immunosuppression and tumor progression. The identification of metabolism-related genes (MRGs) associated with immune reactions in pancreatic cancer may lead to improved treatments. This review highlights the characteristics of MRGs in pancreatic cancer and suggests that enhanced anti-cancer therapies could be used to overcome resistance to immunotherapy.
Pancreatic cancer is the fourth leading cause of cancer-related deaths worldwide, with a five-year overall survival rate of less than 8% [1]. It typically arises in the pancreatic duct, and the known risk factors for pancreatic ductal adenocarcinoma (PDAC) include smoking, obesity, and diabetes. The incidence of PDAC is higher in women than men. Excessive alcohol intake and a family history of PDAC may also contribute to increased risk [2, 3, 4, 5, 6]. Patients with pancreatic cancer often present with advanced disease due to the lack of early detection [7, 8, 9]. Although surgery remains the best treatment option, PDAC is often discovered at an advanced stage when lesions cannot be removed. Consequently, a multidisciplinary therapeutic approach has been implemented for PDAC [8]. Adjuvant chemotherapy has improved long-term outcomes in patients with advanced cancer, although the response rates remain low. Up to 80% of patients with pancreatic cancer have a poor initial prognosis [10]. Even patients who undergo successful surgery eventually develop local recurrence or metastasis, which then greatly worsens the disease prognosis [11]. Although successful in some cancer types, immunotherapy has not been effective in the treatment of pancreatic cancer, with most clinical trials for PDAC giving negative results [8]. Therefore, alternative therapeutic approaches are urgently required for PDAC.
Metabolic reprogramming determines the function and viability of cancer cells and plays an important role in tumor initiation and progression, including PDAC [12, 13, 14]. Tumors undergo dramatic changes in glucose, lipid, and amino acid metabolism compared with normal tissues [15]. Growing evidence has shown the metabolic requirements of immune cells in the tumor microenvironment (TME) have a significant impact on the success of immunotherapy [12, 16]. Despite the presence of many immune cells in pancreatic cancer tissue, immune dysfunction is observed when the TME is immunosuppressive, thereby hindering the activation of immune effectors [17]. In order to develop effective treatment strategies, it is therefore critical to uncover the mechanisms that underlie abnormal metabolism in cancer [15].
Glucose metabolism has been implicated in cancer development and metastasis and is the primary source of carbon chains for biosynthesis and energy metabolism [18]. In normal cells, glucose metabolism involves the synthesis of adenosine triphosphate (ATP) through anaerobic glycolysis and cellular respiration. However, glucose metabolism in cancer cells is aerobic and is regulated by epigenetic mechanisms and cooperation with the TME, which can lead to the loss of tumor suppressors [19, 20]. The rapid proliferation of cancer cells is facilitated by lactate production and glucose metabolism, which lead to cancer progression, chemoresistance, and local depletion of oxygen. This results in hypoxic regions containing high levels of lactate [21, 22, 23], which then restricts immune cell function by switching to an acidic environment [24]. Pancreatic cancers show upregulated expression of rate-limiting glycolytic enzymes including hexokinase 1/2 (HK1/2), phosphofructokinase 1 (PFK1), and lactate dehydrogenase A (LDHA, a subunit of LDH), thus contributing to the Warburg effect and improving lactate-mediated glycolysis [22].
Non-glucose-mediated metabolism, such as amino acids and lipids, is also necessary for cancer cell survival and growth. The main function of amino acids in normal cells is the synthesis of new proteins, and amino acid metabolism in cancer cells is also an essential factor for cell proliferation [25]. Glutamine is important for the production of metabolic intermediates, energy, and cellular functions [26, 27]. Importantly, dysregulation of glutamine metabolism in the tricarboxylic acid (TCA) cycle, the central metabolic hub of cells, is an important driver of cancer cells [28]. Cancer cell growth and proliferation depend on TCA cycle intermediates and amino acids to generate purine and pyrimidine nucleotides [18]. Amino acids are involved in biosynthesis and in the maintenance of redox balance, as well as contributing to immune responses associated with tumor development and metastasis via epigenetic regulation [29].
Lipids are required for membrane biosynthesis during rapid cell proliferation. In addition, they serve as energy stores during metabolic stress, and participate in various signaling pathways [21]. While most normal cells preferentially use extracellular lipids to synthesize new structural lipids, cancer cells are able to synthesize fatty acids (FAs) to maintain proliferation in lipid-poor microenvironments and in the absence of extracellular lipids [30]. Lipid metabolism is reprogrammed in cancer cells and contributes to rapid tumor growth through increased lipid uptake, storage, and lipogenesis [31, 32, 33, 34]. These observations suggest that targeting of dysregulated lipid metabolism may be a promising approach for cancer treatment.
Cell metabolism and metabolites were recently reported to be crucial regulators of the immune system. Therefore, understanding the differential metabolic requirements of various cell types provides an opportunity to highlight metabolic vulnerabilities and expose therapeutic windows that may improve immunotherapy [13]. Immunotherapy is currently shifting the paradigm of cancer therapy. It is important to target metabolism in the normal immune system when treating cancer, particularly for solid tumors such as pancreatic cancer which often contain more non-cancer cells than cancer cells [18].
Glycolysis is a vital metabolic process that produces ATP from glucose to maintain cellular growth. Pancreatic cancers exhibit dysregulated cellular metabolism, which is one of the hallmarks of cancer (Fig. 1) [9]. The Warburg effect is a representative feature of this rewired glucose metabolism that provides adequate ATP for tumor cells to grow, proliferate, and survive [35]. Anaerobic glycolysis is preferred even under normoxic conditions in cancer cells, as it allows the rapid production of metabolites [36]. Mutations in the Kristen Rat Sarcoma Viral oncogene homolog (KRAS) and its downstream pathways regulate glucose metabolism-related genes (MRGs) in PDAC, leading to an increased need for glucose and modulation of the TME [8]. As an example, the rate-limiting enzymes and glucose transporters in glycolysis, PFK1, HK2, and LDHA, are all controlled by oncogenic KRAS [9].
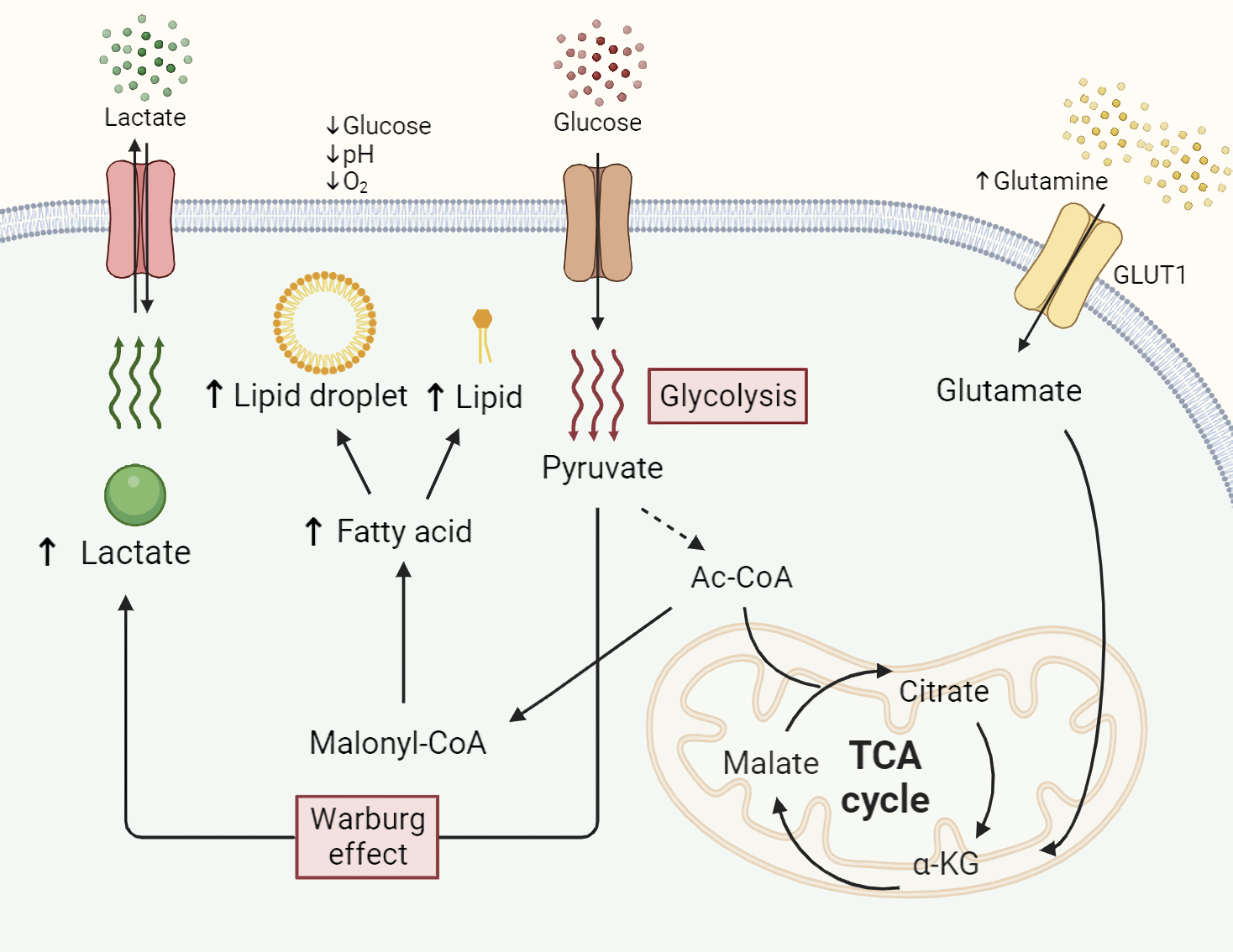 Fig. 1.
Fig. 1.Altered metabolism of glucose, glutamine, and lipids in
pancreatic cancer. Abnormal metabolism of essential nutrients is a hallmark
feature in cancer cells compared to normal cells. Enhanced uptake of glucose in
cancer preferentially undergoes anaerobic glycolysis (Warburg effect), resulting
in elevated lactate production to generate more ATP. Increased influx of
glutamine is facilitated by metabolism-related genes (MRGs) and participates in
the tricarboxylic acid (TCA) cycle by undergoing conversion into glutamate and
Resistance to immunotherapy remains a major challenge in PDAC treatment, despite extensive investigations with this anti-cancer therapy. Metabolic interactions with the immune response may provide some clues to address this limitation. In this regard, reprogrammed metabolic processes in pancreatic cancer have been shown to influence immune cells in the TME, resulting in tumor immunosuppression and resistance to immunotherapy [37]. Since glucose is continuously consumed by the Warburg effect, immune cells compete with cancer cells for nutrition [37]. Moreover, elevated levels of glycolysis induce acidic conditions in the TME due to the accumulated lactate. This allows cancer cells to avoid immune surveillance, but also debilitates the immune response [38]. Lactate accumulation in PDAC reduces the efficacy of immunotherapy by creating an immunosuppressive microenvironment. T cell activation is especially important for functional immunity and is impacted by metabolic changes in the TME. Due to the massive uptake of glucose by cancer cells, the level becomes insufficient for T cell activation [27]. This leads to T cell exhaustion, which inhibits appropriate binding between the immune checkpoint and its ligands in PDAC [39]. As a consequence, the elevated glucose metabolism in PDAC results in immune evasion.
Several MRGs form a vital link between glucose metabolism and the immune response in PDAC. Glucose transporter 1 (GLUT1) mainly contributes to the glycolytic process as a membrane protein. It is highly upregulated in many cancer cell types due to their increased demand for glucose [40, 41]. The crucial role of GLUT1 in generating ATP from glucose makes it a potential biomarker for pancreatic cancer and a target for therapy. Immune cells such as T cells, B cells and macrophages, as well as tumor cells require considerable amounts of energy for optimal function [42]. A glucose-depleted TME can therefore suppress T cells [43]. GLUT1 also participates in immune escape by PDAC through a positive relationship with programmed cell death ligand 1 (PD-L1) and an inverse relationship with programmed cell death 1 (PD-1), both of which are immune checkpoints [41]. Moreover, binding of PD-1/PD-L1 in the TME inversely hinders T cell function, leading to immune evasion [44].
HK2 is a key enzyme in the first step of glycolysis and is highly expressed in most tumors [45]. Together with GLUT1, increased HK2 expression supports the Warburg effect. It serves as an altered glycolytic process and produces sufficient ATP in the TME even under hypoxic conditions [46]. Furthermore, patients with high HK2 expression show anchorage-independent cell growth, metastatic signatures, and poor overall survival [47, 48]. HK2 is negatively associated with immune processes in PDAC. Elevated glycolysis by HK2 generates lactate and glucose, resulting in immunosuppression. Lactic acid can also be utilized by cancer cells as a nutrient substitute [49]. Acidification of the TME by accumulated lactate can suppress cytotoxic T cells and natural killer (NK) cells, which are vital for impeding tumorigenesis [49].
Insulin-like growth factor-binding proteins (IGFBPs) respond to insulin signaling by binding and stabilizing insulin-like growth factor (IGF). Of note, the expression of IGF receptor (IGFR) is elevated in severe-grade tumors such as PDAC [50]. Insulin is produced in the pancreas and transforms glucose into the glycogen storage form. It is therefore strongly associated with both glucose metabolism and pancreatic cancer. IGFBP-1/2 can be downregulated by insulin to activate free IGF, thereby promoting cancer progression [51]. IGF-1 modulates apoptosis, proliferation, metastasis, and the immune system [50, 51, 52]. It also allows CCL5 secreted from peripheral cells to gather in anti-tumor immune cells and tumor-associated M2 macrophages. Moreover, cancer-associated fibroblasts (CAFs) in PDAC can produce IGF ligands [50, 53]. IGFBP-6 plays a role in immunosuppression and chemoresistance, both of which are related to T cell malfunction [54]. Thus, IGFBPs could be used as prognostic biomarkers and therapeutic targets in pancreatic cancer.
The enzyme aldehyde dehydrogenase 1 family member A3 (ALDH1A3) alters the glucose flux and is associated with aggressive PDAC [55]. It also increases HK2 expression, leading to enhance glycolysis [56]. ALDH1A3 is expressed in cancer stem cells (CSCs), which can initiate cancer through differentiation and self-renewal [57]. Elevated levels of ALDH1A3 therefore indicate stemness features of pancreatic cancer, including metastasis and progression [58]. CSCs participate in the immunosuppressive TME as ‘latency competent’ CSCs, which refers to their ability to decrease NK cells while transitioning into a dormant state and evading cytolysis [59]. The plasticity of CSCs in PDAC allows them to escape immunotherapy because they persist for extended periods and are promoted by hyperglycemia [60].
Aldolase A (ALDOA) catalyzes fructose 1,6-bisphosphate into glyceraldehyde
3-phosphate (G3P) and dihydroxyacetone phosphate in the glucose metabolic pathway
[61]. This enzyme is highly expressed in most cancer types. In pancreatic cancer,
ALDOA is associated with malignant characteristics such as cell proliferation,
invasion and migration, and worse patient survival [40, 62]. ALDOA is thus
closely associated with PDAC metastasis and progression. Silencing of ALDOA in
PDAC cell has been shown to downregulate crucial molecules in glycolysis,
including hypoxia-inducible factor 1 (HIF-1) [63]. Moreover, ALDOA is associated
with HIF-1-related genes and reduces CD8
Amino acids are crucial nutrients for most cells, including cancer cells, with
glutamine being the main reprogrammed amino acid in cancer [66]. Glutamine has
multifaceted functions that affect PDAC progression. It can support the TCA cycle
by converting to
Given its multiple roles, glutamine is able to coordinate various immune reactions. A sophisticated TME organized by dysregulated glutamine metabolism can attenuate anti-tumor immune processes in PDAC [72]. T cell proliferation is enhanced by increasing the levels of glutamine transporters, glutaminolysis-associated enzymes, and Myc transcription factor via T cell receptor (TCR)-stimulated activation [73]. Elevated expression of these proteins can meet the demands of anabolic mechanisms and immune cells [73]. However, excessive glutamine consumption by cancer cells interferes with the appropriate utilization of glutamine by immune cells, leading to the reprogramming of immune cell metabolism [74]. This affects tumor immune cell infiltration by inhibiting T cell activation through the upregulation of PD-L1 expression [73, 75]. Hence, the targeting of PD-L1 in combination with glutamine MRGs may address the challenges associated with immune checkpoint blockade.
Transcription factor EB (TFEB) is a critical molecule in autophagy and lysosomal function. It forms part of the coordinated lysosomal expression and regulation (CLEAR) gene network response to nutrient availability, including amino acids [76]. In PDAC, TFEB shows increased expression following glutamine metabolism [77]. TFEB regulates the cellular process of autophagy, which is responsible for the degradation and recycling of intracellular components. Autophagy plays a vital role in cancer cells by providing alternative energy sources for cell growth. Since cancer cells utilize glutamine and TFEB modulates glutaminase expression, the regulation of TFEB is a significant factor in PDAC growth [77]. Moreover, TFEB is linked to innate immunity and inflammation through its activation of macrophages and promotion of autophagy [78]. TFEB can also enhance PD-L1 expression and help cancer cells evade immune responses by inhibiting mammalian target of rapamycin (mTOR) [79].
Metabolic pathways for cellular biosynthetic sources interact with various metabolic genes. For example, glutamine-fructose-6-phosphate transaminase 2 (GFPT2) is a rate-limiting enzyme in the hexosamine biosynthesis pathway (HBP) that promotes glucose uptake in cancer cells [80]. Increased GFPT2 expression in hypoxic pancreatic cancer is associated with sufficient access to both glucose and glutamine via the HBP [37]. GFPT2 favors the epithelial-mesenchymal transition (EMT) and cell invasion and migration, leading to poor prognosis [80, 81]. Additionally, GFPT2 is closely associated with immune infiltration in the TME, resulting in the progression of cancer through the release of growth factors from immune cells, including T cells, B cells, and macrophages [82]. Interestingly, GFPT2 is also linked to T cell exhaustion and immunosuppressive factors, such as macrophages and fibroblasts. This occurs via the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway and infiltrated immune cells [83]. Thus, GFPT2 is a promising MRG for the targeting of immune escape in cancer.
Excitatory amino acid transporters (EAATs) can maintain the glutamate status and
transform glutamine-derived glutamate into
The L-type amino acid transporter LAT1, also known as SLC7A5, is essential for
metabolism during the progression of cancer through the provision of sufficient
amino acids [88]. LAT1 is able to transport amino acids even under conditions of
low affinity, and can fully activate CD8
Lipids are an essential nutrient source for cancer and normal cells. In cancer cells, lipids provide fuel for cell membranes, ATP production, and signaling pathways [92]. Therefore, altered lipid metabolic processes contribute to the metastasis, invasion, and proliferation of tumors [93]. Increased de novo lipid synthesis leads to the accumulation of fatty acids (FAs), resulting in tumor progression [94]. KRAS mutations occur in most pancreatic cancers and have been shown to influence FA oxidation and lipid droplet (LD) storage by repressing hormone-sensitive lipase (HSL), thereby favoring PDAC cell invasion and migration [94]. Because the requirement for lipids is elevated in advanced pancreatic cancer, the lipolysis-related enzymes fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC) are upregulated [95]. In addition, an abundance of lipids and immune complexes can lead to inflammatory reactions [33].
A deeper understanding of the connection between immune responses and metabolic processes is required to fully appreciate the impact of the immune system on cancer cells (Fig. 2). Enhanced lipid metabolism leads to greater demand for lipids, FA oxidation and FAS, and results in the creation of an immunosuppressive niche [96]. While lipolysis exhibits a tendency converse to lipogenesis to support excessive provision of lipids. Downregulation of HSL suppresses the breakdown of stored lipids and triggers PDAC metastasis [97]. Furthermore, reprogrammed lipid metabolism in tumor-associated macrophages (TAM) in the TME helps to regulate cancer immunity [98]. Lipids accumulate in TAM and promote M2 macrophage traits [99]. Lipid metabolism subsequently releases anti-inflammatory chemokines and cytokines that attract regulatory T (Treg) cells and present the PD-L1 and CD80 ligands to the immune checkpoint inhibitors PD-1 and cytotoxic T-lymphocyte-associated antigen 4 (CTLA4). This leads to diminished anti-cancer immunity related to T cell function [98, 100]. Accordingly, abnormal lipid metabolism is relevant not only for the immune response in the TME, but also as a target of immunotherapy resistance.
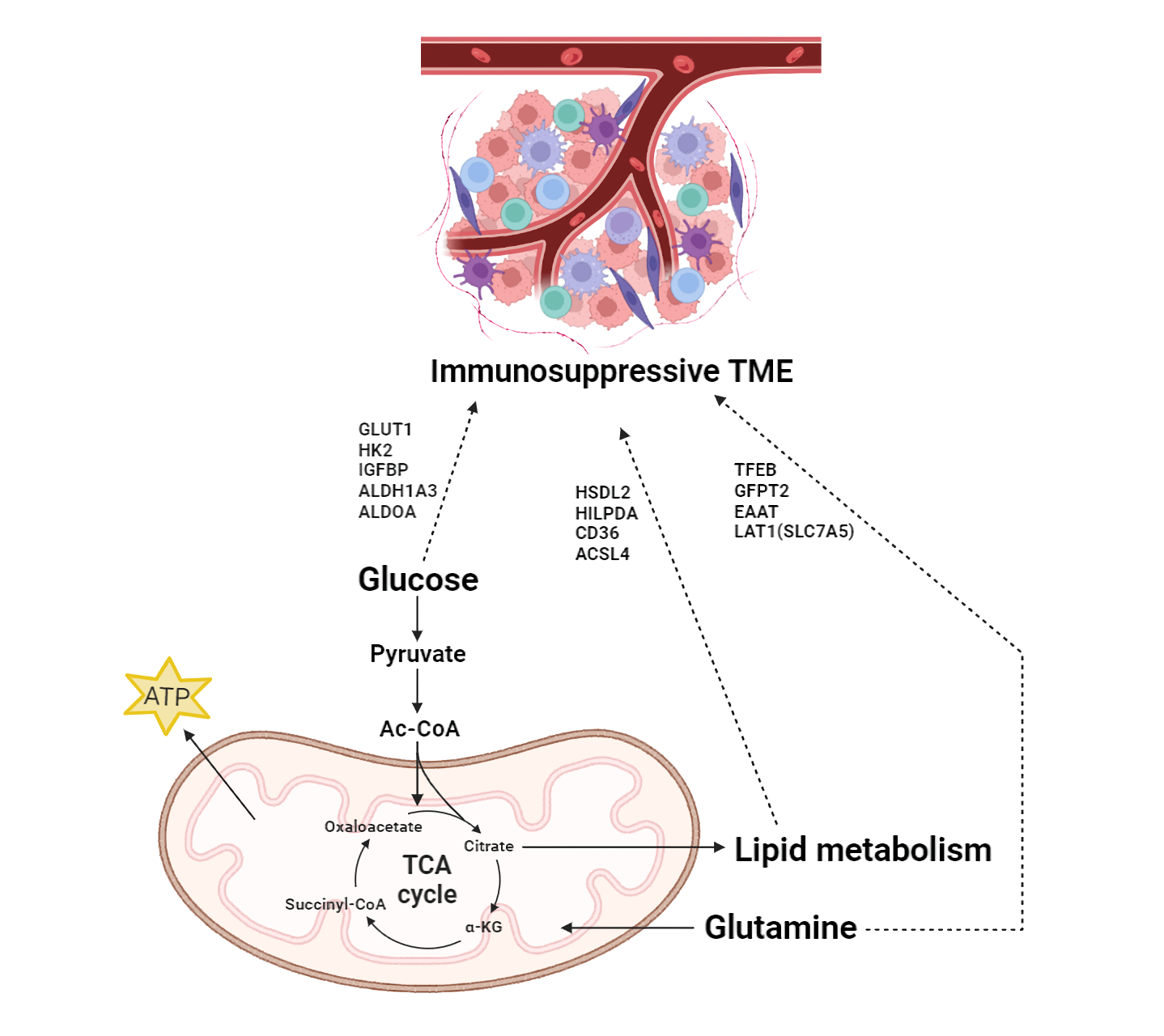 Fig. 2.
Fig. 2.Impact of MRGs on the immunosuppressive Tumor microenvironment
(TME) in pancreatic cancer. Glucose, glutamine, and lipid metabolism contribute
to cancer cells through the TCA cycle. They are connected to cellular energy
production and synthesis, thereby influencing the overall cellular metabolic
landscape and supporting the biosynthetic demands of rapidly proliferating cancer
cells. Moreover, MRGs induce an immunosuppressive environment in the TME and
connect metabolism with the immune response in PDAC. TME, tumor microenvironment;
GLUT1, glucose transporter 1; HK2, hexokinase 2; IGFBPs, insulin-like growth
factor binding proteins; ALDH1A3, Aldehyde dehydrogenase 1 family member A3;
ALDOA, Aldolase A; TFEB, Transcription factor EB; GFPT2,
Glutamine-fructose-6-phosphate transaminase 2; EAAT, Excitatory amino acid
transporters; LAT1, L-type amino acid transporter 1; HSDL2, Hydroxysteroid
dehydrogenase like protein 2; HILPDA, Hypoxia inducible lipid droplet associated;
ACSL4, Long chain acyl CoA synthetase 4; Ac-CoA, acetyl CoA;
Hydroxysteroid dehydrogenase-like protein 2 (HSDL2) is a significant FA enzyme in lipid metabolic pathways relevant to tumorigenesis [101]. The expression of HSDL2 is increased in PDAC compared to adjacent tissues and is associated with poor prognosis [101, 102]. HSDL2 also promotes the first stage of cancer progression involving increased cell proliferation and migration via EMT [102, 103]. Moreover, HSDL2 interacts with peroxisome proliferator-activated receptors (PPARs) to induce Tregs and immunosuppressive activity, which are crucial for regulating innate immune responses and inflammation [104]. HSDL2 has drawn attention as a candidate gene for immunosuppression in pancreatic cancer treatment due to its association with immune cell infiltration.
Hypoxia-inducible lipid droplet-associated (HILPDA) reacts to oxygen or glucose starvation in PDAC and macrophages by increasing the capacity of LDs to store neutral lipids [105]. The modulation of triglyceride and LD development through attachment and prevention of Adipose Triglyceride Lipase (ATGL) activity, a rate-limiting lipase, is critical for tumor growth [105]. HILPDA is highly expressed in cancer cells, and increased lipid metabolic processes result in the accumulation of incoming lipids into LDs. Consequently, HILPDA expression tends to promote tumor growth, leading to poor survival of pancreatic cancer patients [106]. HILPDA expression correlates with infiltrated immune cells, such as TAM, and also with immunosuppressive genes, including PD-L1/PD-1 [107]. Abnormal HILPDA function in cancer cells may therefore impede the efficacy of anti-cancer treatment.
CD36 contributes to FA uptake under dysregulated metabolic conditions, especially under high lipid requirements [108]. CD36 establishes lipid homeostasis as an FA transporter and accelerates the progression and metastasis of pancreatic cancer [109]. Interestingly, CD36 expression in PDAC is lower than in normal tissues as an unfavorable prognostic marker but shows shorter overall survival, indicating a more severe tumor [109, 110]. Downregulated CD36 expression reduces cell adhesion to the Extracellular matrix (ECM) and induces cell motility, resulting in increased metastasis [109]. Additionally, CD36 expression is associated with immune cell infiltration and with checkpoint inhibitors involving PD-L1 [111]. Along with CD36, lipids can also regulate immunity in cancer cells, and altered lipid metabolism leads to both tumorigenesis and immune tolerance [111].
Long-chain acyl-CoA synthetase 4 (ACSL4) controls de novo lipogenesis
by increasing the levels of lipogenic regulators in cancer cells [112]. ACSL4
expression in cancer cells leads to the accumulation of lipids from aberrant
metabolic states. This occurs sequentially via increased levels of Myc, sterol
regulatory element binding protein 1 (SREBP1), and FA synthesis, which then
eventually induces cancer cell growth and metastasis [112]. Furthermore, ACSL4
induces an immunosuppressive TME in PDAC, resulting in poor prognosis [113].
ACSL4 is also associated with ferroptosis, a form of regulated cell death, and
its downregulation promotes cancer progression [113]. This process functions in
an anti-tumor immune response, followed by CD8
The significant progress in tumor biology has led to new methods for treating multifarious cancers. Treatment with immune checkpoint inhibitors has been widely used as a novel strategy, but has significant limitations. Abnormal metabolic changes are a distinct feature of malignant cells, and hence the ability to regulate cell metabolism may give rise to new treatment options (Fig. 3).
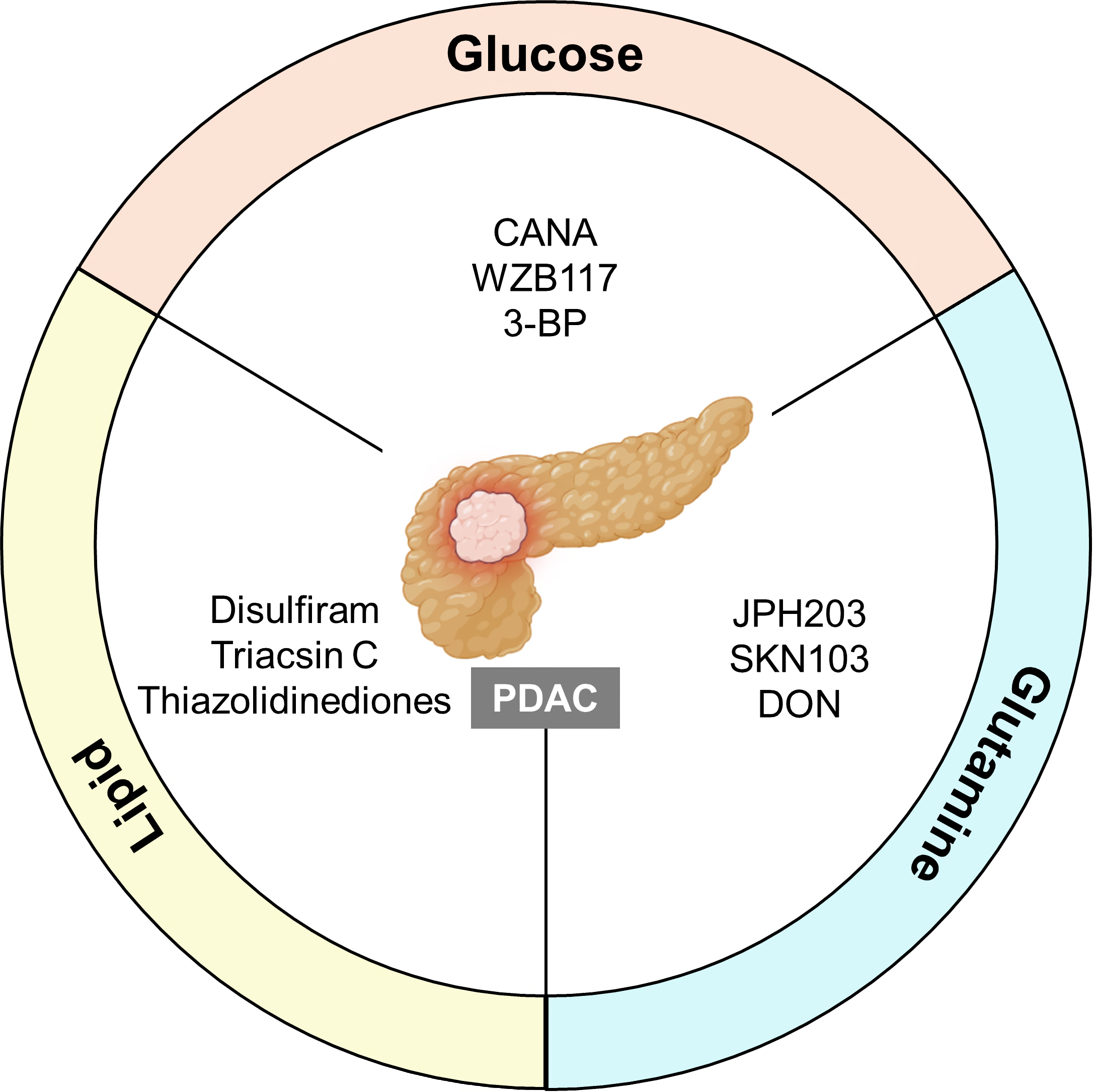 Fig. 3.
Fig. 3.Implications of MRGs in pancreatic cancer. The targeting of glucose, glutamine, and lipid MRGs may be a promising strategy to treat PDAC. Each metabolic pathway can promote or suppress crucial functions in cancer cells to modulate tumor development and progression. Inhibitors mainly regulate the major MRGs in metabolic processes and exacerbate immunosuppression. Hence, they could also be utilized in combination therapy. PDAC, pancreatic ductal adenocarcinoma; CANA, canagliflozin; 3-BP, 3-bromopyruvate; DON, 6-diazo-5-oxo-L-norleucine.
Modulation of glucose uptake is important for metabolism-associated treatment. Of the genes mentioned earlier, GLUT1 is a well-known enzyme involved in glucose metabolism and is currently the subject of active research. Canagliflozin (CANA) is an anti-diabetic drug that acts by inhibiting sodium-glucose cotransporter 2 (SGLT2), which manages glucose flux. CANA has reportedly shown efficacy against hepatocellular carcinoma (HCC) [115] and can reduce the viability and growth of cancer cells through necrosis, both in vitro and in vivo [116, 117]. CANA can promote apoptosis and suppress glycolysis in pancreatic cancer by decreasing GLUT1 expression through the phosphoinositide 3 kinase/AKT/mTOR (PI3K/AKT/mTOR) pathway [117]. CANA also improves the efficacy of the chemotherapeutic drug gemcitabine, indicating a synergetic effect when used in combination with current medication [117, 118]. WZB117 is a competitive GLUT1 inhibitor that targets glycolysis. However, these inhibitors are still undergoing Phase II clinical trials [47, 119]. WZB117 decreases cancer progression in vivo by reducing ATP and glycolytic enzyme levels in a dose-dependent manner [116]. Combination therapy with GLUT inhibitors and immunotherapy appears to weaken tumor cell lysis and overcome resistance [120].
Given that glutamine contributes to tumor growth and survival, targeted therapy against glutamine MRGs holds promise for overcoming resistance to immunotherapy. The LAT1 inhibitor JPH203 has completed first-in-human Phase I trials and is currently undergoing randomized Phase II clinical trials [121, 122]. JPH203 prevents the in vitro and in vivo progression of PDAC by blocking transport of bulky neutral amino acids [122, 123]. SKN103 is another LAT1 inhibitor that can inhibit pancreatic cancer cell growth in vitro [124]. Moreover, the glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) enhances the efficacy of intra-tumoral MBTA immunotherapy [125]. MBTA refers to the combination of Mannan-BAM, Toll-like receptor (TLR) ligands, and Anti-CD40 antibody. This immunotherapy can activate not only innate immunity but also the adaptive immune response in pancreatic cancer [126]. Therefore, DON can sensitize PDAC to immunotherapy within the TME [125].
Cancer cells facilitate the production of lipids, glucose, and glutamine. This has led to the suggestion of novel therapeutic approaches for pancreatic cancer that involve lipid-related MRGs. Lipids mediate the immune system in the TME in several ways. Cyclooxygenase-2 (COX-2) inhibitors favor the immune response by decreasing myeloid-derived suppressor cells (MDSCs). This results in a permissive TME for T cells rather than the immunosuppressive TME in pancreatic cancer [127]. COX-2 inhibitors can potentially be co-administered with immune checkpoint blockers to reduce the adverse effects of immunotherapy. Combination treatment with COX-2 inhibitor and different drugs, such as celecoxib and aspirin, is still undergoing clinical trials in various cancer types [127]. Disulfiram targets aldehyde dehydrogenase (ALDH), which is involved in both lipid and glutamine metabolism, and induces anti-tumor activity in pancreatic cancer both in vitro and in vivo [128, 129, 130]. In particular, disulfiram inhibits PDAC growth by suppressing cell proliferation [128]. This suggests that combined treatment with MRGs may be an effective anti-cancer therapy to address the current challenge of immunotherapy resistance. Because aberrant metabolism mostly results in immunosuppression of the TME, targeting these MRGs has the potential to increase the efficacy of pancreatic cancer treatment (Table 1, Ref. [116, 117, 122, 131, 132, 133, 134, 135, 136, 137]).
| Related metabolism | Cellular target | Drug | Function | FDA-approval | Reference |
| Glucose | GLUT1 | CANA | Enhances apoptosis and inhibits glycolysis | Yes | [117] |
| WZB117 | Suppresses cancer growth | No | [116] | ||
| HK2 | 3-BP | Restrains ATP generation and induces apoptosis in cancer | No | [131] | |
| Glutamine | LAT1 | JPH203 | Represses PDAC progression by obstructing amino acids transporters | No | [122] |
| SKN103 | Downregulates cancer development | No | [132] | ||
| Glutaminase | DON | Inactivates tumor metabolism and alleviates hypoxia | No | [133] | |
| Lipid | ALDH | Disulfiram | Upregulates anti-tumor effect | Yes | [134] |
| ACSL4 | Triacsin C | Arrests ACSL4 via competing with FAs and induces ferroptosis | No | [135, 136] | |
| Thiazolidinediones | Suppresses ACSL4 and reduces cancer cell growth | No | [136, 137] |
GLUT1, glucose transporter 1; CANA, canagliflozin; HK2, hexokinase 2; 3-BP, 3-bromopyruvate; ATP, adenosine triphosphate; LAT1, L-type amino acid transporter 1; PDAC, pancreatic ductal adenocarcinoma; DON, 6-diazo-5-oxo-L-norleucine; ALDH, aldehyde dehydrogenase; ACSL4, long chain acyl CoA synthetase 4; FAs, fatty acids; FDA, U.S. Food and Drug Administration.
Although the targeting of specific metabolites may be a promising treatment approach, several concerns remain. Cancer cells exhibit considerable dependence on nutrients for their growth. Several metabolic pathways in tumor cells function in tandem with the surrounding cells [138]. Therefore, metabolic changes that occur after targeting MRGs might lead to unwanted interactions between the heterogeneous TME. In other words, changes to MRGs only may result in unsatisfactory outcomes in PDAC patients, and application of combination therapy may be required. Currently, clinical trials of combination therapies with simvastatin, digoxin and metformin aim to assess their therapeutic efficacy in PDAC and other advanced solid tumors [8]. Future studies aimed at developing metabolism-related strategies must overcome conventional obstacles, including immunosuppression. This review provides some useful insights for the investigation of MRGs as possible targets.
The alteration of various metabolites, including glucose, amino acids, and lipids, is a conspicuous feature of cancer progression. Cancer cells consume essential nutrients to fuel the excessive growth of tumors and to modify the TME in order to survive. Immunotherapy, primarily against the immune checkpoints PD-L1/PD-1 and CTLA4, is used in pancreatic cancer treatment. Although various remedies to cure cancer have been investigated, the outcome of PDAC exhibits little promise in terms of the detrimental effects of immunotherapy. Targeted therapies against MRGs may overcome this limitation, and combination treatments with existing strategies could result in improved outcomes. Numerous studies have demonstrated the potential of MRG-associated therapies for pancreatic cancer. Recent developments with single cell analysis platforms allow the study of targeted genes in the PDAC phenotype [139, 140]. However, clinical trials have yet to be performed, and it is uncertain whether anti-cancer treatments using MRG will be beneficial. In view of the complexity of abnormal metabolic processes and their interactions within the TME, further careful research is necessary. Resistance to immunotherapy is a critical barrier that must be overcome to obtain effective anti-tumor treatments in various cancer types. MRGs have been identified as significant therapeutic targets and as potentially useful in combination therapies for PDAC.
SC, WK, EK, SS, MS, HJK, and HY conceptualized the study. SC, WK, EK, SS, MS, HJK, and HY prepared the original draft of the manuscript. SC, WK, EK, SS, MS, HJK, and HY reviewed and edited the manuscript. HY supervised the study. HY was responsible for funding acquisition. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors wish to acknowledge the financial support of the Catholic Medical Center Research Foundation made in the program year of 2019.
The authors wish to acknowledge the financial support of the Catholic Medical Center Research Foundation made in the program year of 2019, and funded by the Brain Korea 21 (BK21), grant number M2022B002600003, and the Ministry of Food and Drug Safety in Korea, grant number 22213MFDS421.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.