
- Academic Editor
Sodium-glucose cotransporters 2 (SGLT2) are high-capacity, low-affinity transporters, expressed mainly in the early portion of the proximal renal tube, mediating up to 90% of renal glucose uptake, while SGLT1 receptors are found mainly in the small intestine, facilitating glucose absorption. SGLT2 inhibitors (SGLT2i) originally emerged as agents for the treatment of type 2 diabetes mellitus; however, they soon demonstrated remarkable cardio- and renoprotective actions that led to their licensed use for the treatment of heart failure and chronic kidney disease, regardless of the diabetic status. Cardiovascular remodelling represents an umbrella term that encompasses changes that occur in the cardiovascular system, from the molecular and cellular level, to tissue and organs after local injury, chronic stress, or pressure. SGLT modulation has been shown to positively affect many of these molecular and cellular changes observed during pathological remodelling. Among the different pathophysiological mechanisms that contribute to adverse remodelling, various stem and progenitor cells have been shown to be involved, through alterations in their number or function. Recent studies have examined the effects of SGLT2i on stem and progenitor cell populations and more specifically on endothelial progenitor cells (EPCs). Although some found no significant effect, others showed that SGLT2i can modulate the morphology and function of EPCs. These preliminary observations of the effect of SGLT2i on EPCs may be responsible for some of the beneficial effects of gliflozins on pathological remodelling and, by extension, on cardiovascular disease. The purpose of this narrative review is to critically discuss recent evidence on the cardioprotective effects of SGLT2is, in the context of cardiac remodelling.
Cardiac remodelling is a complex biological process, characterized by a number of changes in molecular and cellular tissue architecture, in response to long-term stress, such as pressure or volume overload, tissue inflammation, or local ischemia due to myocardial infarction; gross tissue function will eventually change as well, to accommodate these new conditions. Remodelling may be a physiological adaptation, for example, during exercise or pregnancy, or it may be pathological, for example following chronic pressure overload or ischemic injury. A number of different mechanisms have been discovered to contribute to this process, from alterations in cellular and subcellular organization, to metabolic derangements, inflammation, and even stem and progenitor cell dysfunction [1, 2].
Eventually, the resulting cardiac dysfunction can lead to heart failure (HF) [1], often described as a clinical syndrome [3]; in fact, in 2019, the Heart Failure Association (HFA) ATLAS Project estimated the median prevalence of HF to be 17 per 1000 persons, while in North America the prevalence is projected to increase from 2.4% (2012) to an estimated 3.0% in 2030 (United States). On the other hand, incidence is relatively stable and has been shown to be decreasing, at least in the developed world [4].
Risk for HF may increase due to various conditions, including hypertension and coronary artery disease; type 2 diabetes mellitus (T2DM) in particular, confers an increased risk of HF-related morbidity and mortality, along with an increased risk of occurrence, highlighting the need for prompt and appropriate management [5]. Not only do therapies for both HF and diabetes mellitus act synergistically, but treatment of one disorder can positively affect the outcomes in the other [6]. Among the various pharmaceutical compounds developed for the treatment of T2DM, sodium-glucose cotransporters 2 inhibitors (SGLT2is) have emerged as a viable option, due to the low risk of hypoglycemia as well as the weight-lowering effects [7]. They were first studied more than 20 years ago, when their potential for inhibition of the kidney SGLT transporter was evaluated [8]. Ever since, their effectiveness in the treatment of hyperglycemia [9], mitigation of kidney damage [10] and alleviation of cardiovascular risk has been confirmed [5]. SGLT inhibition has been shown to exert protective effects, not only on kidney function, but also on the heart and vasculature [11]. Recent research on SGLT2is has revealed a variety of mechanisms, targeting several of the derangements observed in pathological remodelling [5].
Although there has been extensive research into the mechanisms that could contribute to pathological remodelling, evidence for stem and progenitor cell involvement is relatively limited. In more detail, dysfunctional or even decreased numbers of progenitor populations have been shown to be involved in the process [12, 13]. Investigation into the relationship between SGLT modulation and progenitor cell function has been a relatively recent pursuit, with few studies examining this association yet, often yielding conflicting results [14]. Thus, the purpose of this narrative review is to gather and critically discuss recent evidence on the cardioprotective effects of gliflozins, in the context of cardiac remodelling, with an emphasis on pathological remodeling.
SGLTs are a large family of membrane cotransporters found on the plasma membrane
of various cells that, in addition to glucose, transport various other molecules,
such as vitamins, amino acids, and ions across the apical cellular membrane. In
general, there are 6 SGLT isoforms, of which SGLT1 and SGLT2 have been the most
studied. SGLT1 is most frequently found in the small intestine and is responsible
for glucose and galactose absorption. The first SGLT protein transporter to be
discovered [7], SGLT1 is encoded by the SLC5A1 gene and consists of 664
amino acids. It functions primarily by facilitating the simultaneous transport of
2 Na
The series of structural and functional changes observed in cardiac remodelling assist to the better adjustment of the heart to new conditions, leading to either physiological or pathological adaptation; these may be caused by a variety of different factors which are summarized in Table 1 [18, 19, 20, 21, 22, 23, 24, 25, 26]. Eventually, left ventricle (LV) pressure, radius, and wall thickness are altered, so that the developed wall stress remains within an appropriate range to maintain the cardiac output (CO) [19].
| Physiological remodelling | Normal growth (neonatal/postnatal period) |
| Normal aging | |
| Exercise | |
| Pregnancy | |
| Pathological remodelling | Pathological aging-related processes |
| Inflammation | |
| Infiltrative conditions (cardiac amyloidosis, sarcoidosis, hemochromatosis) | |
| Autoimmune conditions (RA, SLE, PM/DM, pSS, SSc) | |
| Ischemia | |
| DCM | |
| Endocrine conditions (for example acromegaly, Cushing’s Syndrome, hyperthyroidism/hypothyroidism, hyperaldosteronism) | |
| HOCM | |
| Chronic mechanical stress | |
| Various drugs/toxins (for example cocaine, amphetamines, isoprenaline, anticancer drugs – doxorubicin, and ethanol, leading to cardiomyopathy) |
RA, Rheumatoid Arthritis; SLE, Systemic lupus erythematosus; PM, polymyositis; DM, dermatomyositis; pSS, primary Sjögren’s Syndrome; SSc, Systemic sclerosis; HOCM, Hypertrophic Cardiomyopathy; DCM, Diabetic Cardiomyopathy.
Changes occurring with physiological remodelling adaptations include alterations in cell morphology, protein expression, and signalling pathways [27]. These frequently result in cardiac hypertrophy through an increase in cardiomyocyte size [19]; a number of signalling pathways are implicated in physiological cardiac hypertrophy, such as mammalian target of rapamycin (mTOR) and phosphoinositide-3-kinase/protein kinase B (PI3K/Akt), leading to increased Akt levels and thus increased cardiomyocyte size [28]. In fact, studies in mice have shown higher levels of PI3K and Akt when cardiomyocytes are subjected to increased pressures (hypertrophy signals), in turn causing an increase in size [29]. The role of Akt in normal growth has been exhibited in Akt-deficient mice; insulin-like growth factor (IGF) or exercise had no effect, which further proved that IGF acts through Akt to induce physiological cardiomyocyte hypertrophy [30]. IGF/Akt is inhibited by SIRT6, proving that SIRT6 is capable of attenuating cardiomyocyte hypertrophy; indeed, SIRT6 has been found in reduced quantities in dysfunctional cardiac tissue and failing hearts, while SIRT6 deficient mice appeared to be unaffected by hypertrophic stimuli in general [31]. Akt may also become activated by mTOR [19], and also lead to cardiomyocyte hypertrophy, experimentally shown through pharmacological mTOR inhibition by rapamycin [32].
Hypertrophy may also be mediated by the expression of certain myosin and
sarcoplasmic reticulum (SR) genes,
| Cardiac remodelling | Signalling mechanisms |
| Physiological | Insulin, PI3K/Akt signalling pathway; Akt interacts with PI3K, affecting L-type calcium channels (LTCC)-Akt inhibited by SIRT6. |
| mTOR/Akt pathway. | |
| TR | |
| Pathological | CM thyroid receptors downregulated (hypothyroid cell state), (pathological aging-induced cardiac remodelling an additional cause); |
| miR-132 overexpression, FoxO3 (Transcription factor-TF) repression and activation of the calcineurin/NFAT signalling pathway, induction of pathological cardiac hypertrophy, impairment of autophagy. |
PI3K, phosphoinositide-3-kinase; Akt, Protein Kinase B; LTCC, L-type Calcium
Channel; SIRT6, Sirtuin 6; mTOR, mammalian target of rapamycin; TR
Pathological cardiac remodelling is characterized by a series of molecular and
cellular changes, related to alterations in cells, subcellular organelles,
intracellular and extracellular protein expression summarized in Table 3, as well
as changes in various intracellular processes, including metabolism, reactive
oxygen species (ROS) and Ca
| Structure | Alteration | Additional information |
| Cardiomyocytes (CM) | Hypertrophy | Increased protein expression (biomechanical stress, neurohumoral activation, fibroblast/myofibroblast growth-factor secretion). |
| Progressive CM lengthening, rearrangement within the myocardial matrix (‘cell slippage’), cell death and replacement by fibrous matrix. | ||
| Cell death | Apoptosis, autophagy, pyroptosis, necroptosis. | |
| Mitochondria | IF mitochondria | IF mitochondria smaller and rounder, increased numbers, increased recruitment of immature mitochondria. |
| Sarcoplasmic reticulum (SR) | Disordered Structure | Changes non-specific, not easily quantified. |
| Cell Membrane | T-tubule | Reduced surface area and density, loss of some T-tubules. |
| Intracellular proteins | Myofibrillar proteins | Increase in absolute numbers, and length, depending on orientation of added sarcomeres (if added in parallel or in series respectively). |
| Remodelling due to pressure increases causes increase in myofibril number, while remodelling due to volume increase causes increase in myofibril length. | ||
| ICD proteins | ICDs exhibit a smaller surface area, along with the presence of vacuoles. | |
| Desmosomes | Increased expression, toxic desmin oligomers. | |
| Extracellular proteins | Collagen | Increased collagen deposition (Type I collagen, type III collagen), mediated by fibroblasts, myofibroblasts, CMs, vascular cells, various immune system cells (macrophages, lymphocytes). |
| Induced by TGF- | ||
| ECM collagen disruption by metalloproteinase activation. |
CM, cardiomyocyte; ICD, intercalated disk; TGF-
| Process | Alteration | Causes/Results |
| Metabolism | Switch towards glycolysis | Increased aspartate production from glucose, low oxygen tension and HIF-1a production also induce switch to glycolysis (cell hypertrophy). |
| Preferential use of ketone bodies (AcAc, D | ||
| Variations in FA metabolism | Increase, decrease, as well as no change in the rate of FA use as substrate; variation in study results due to different stages of remodelling and heart failure studied. | |
| Diminished FA metabolism in later stages of heart failure, may correlate with cardiac function (ejection fraction-EF). | ||
| ROS | ROS increase | Produced by NADPH oxidases (NOX2, NOX4) (NOX2 activation after myocardial infarction), xanthine oxidoreductase, nitric oxide synthase (NOS), by-product of the mitochondrial electron transport chain (ETC). |
| Increase in Na | ||
| ROS from ischemia-reperfusion injury (due to ADP depletion in the early stages, and oxygen reintroduction later on). | ||
| Effects: lipid peroxidation, damage in excitation-contraction coupling proteins, mitochondrial damage, increase in fibroblast (CM hypetrophy through | ||
| TGF- | ||
| TGF- | ||
| Inflammation | Inflammatory pathway activation | Associated with various stages of atherosclerosis (atherogenesis, atherosclerosis and plaque rupture, myocardial infarction). |
| NLRP3 inflammasome formation, resident macrophages and monocytes activation, production of IL-Ib, TNF- | ||
| Epicardial fat deposits secrete factors (IL-1b, IL-6, TNF- | ||
| Ca |
Ca |
Increased Ca |
| Effects: reduction in rate of Ca | ||
| Na |
NKA, phospholemman | Intracellular Na |
| Regulated through phospholemman (inhibits its activity); inhibition of phospholemman mediated through phosphorylation by PKA and PKC. | ||
| ‘Energy-starvation hypothesis’, pathological changes in metabolic processes usually precede changes in contractile function, eventually leading to hypertrophy and heart failure; preferential use of ATP derived from glycolysis for NKA function might explain Na | ||
| Intracellular Na | ||
| SNS | Nerve density and neurotransmitter release | Increased excitability, further increase in neurotransmitter levels (NA) due to circadian rhythm disruption. |
| RAAS | Increased aldosterone-mediated signaling | Aldosterone, through AT1R receptor binding, affects vascular tone and blood volume, may induce fibrotic and hypertrophic changes, increase ROS production, induce inflammation. |
AcAc, acetoacetate; D
Most changes during cardiac remodelling will eventually contribute to increased cardiomyocyte size and cellular death. Cardiomyocyte hypertrophy is often the result of increased protein synthesis and addition of sarcomeres, while it may be attributed to many processes including biomechanical stress, neurohormonal stimulation, and fibroblast, or myofibroblast-derived growth factors [38]. A mechanism described for this process includes progressive lengthening of cardiomyocytes, rearrangement within the myocardial matrix (‘cell slippage’), cell death, and fibrous matrix replacement [39]. Forms of cell death observed in cases of cardiac remodelling include programmed cell death or apoptosis [40], pyroptosis, a form of apoptosis caused by inflammatory processes [41], autophagy, programmed necrotic cardiomyocyte death [39], and regulated necrosis, also known as necroptosis, often observed after myocardial ischemia [42].
During cardiac remodelling a series of alterations in cellular organelles, including mitochondria, the cellular membrane (sarcolemma), and the sarcoplasmic reticulum (SR) have been described. Mitochondria often display abnormal localization, changes in total intracellular mass, or increases in immature forms [43, 44]. Of the various mitochondrial subgroups, only mitochondria located around myofilaments (intrafibrillar-IF) appear to exhibit changes in shape and size, being smaller and rounder than normal, with increased numbers possibly due to increased recruitment of immature forms [43], with the overall area occupied by mitochondria increased as well [45]. The sarcoplasmic reticulum (SR) also seems to exhibit structural changes, with affected cells showing more disordered SR in sheep models of HF, although these changes do not seem to be specific, or easily quantified [45]. Changes in cellular membrane morphology appear to occur as well, particularly in areas where cellular invaginations along the Z-line area of the sarcomere (T-tubules) make contact with the SR [46]. These modifications include reduced surface area and density [47], as well as loss of some T-tubule formations entirely, at least in age-related remodelling [48].
Myofibrillar protein structure is also affected. Depending on the orientation of added sarcomeres, if added in parallel or in series, an increase in absolute numbers, and length have been documented. Remodelling due to elevated pressure usually promotes a proliferation in myofibril number, while remodelling due to volume increase usually leads to an increase in myofibril length [49, 50]. Fetal myosin isoforms may also persist in a greater number in cases of pathological remodelling [1]. Proteins comprising intercalated disks (ICDs) may be affected as well [51], with a higher percentage of ICDs showing a smaller surface area, a mechanism possibly responsible for arrhythmogenicity and conduction disturbances; presence of vacuoles with an increase in associated protein expression may also be observed. Furthermore, desmosome protein expression seems to upregulated, with desmin forming toxic oligomers [52, 53].
The extracellular matrix (ECM) also undergoes changes [54], including both
accumulation and disruption of the normal collagen network, due to
metalloproteinase activation [1]. Collagen accumulation (fibrosis) usually occurs
as a result of tissue injury after myocardial infarction (MI), inflammation or
chronic pressure overload, perpetuated by fibroblasts, myofibroblasts,
cardiomyocytes, vascular cells, and various immune system cells (macrophages,
lymphocytes) [54]. Increased collagen deposition, which can be of type I or III
[20], can be induced by many different signalling pathways, including
TGF-
The heart usually relies on oxidative phosphorylation for about 95% of its
needs in ATP, of which about 6 kg seem to be needed on daily basis by the
myocardium [57]. Glycolysis provides the remaining energy requirements; a number
of substrates are utilized for this purpose, including fatty acids (FA), glucose,
lactate, as well as ketone bodies, such as acetoacetate (AcAc) and
D-
It seems that, in general, other alternative fuels seem to be preferred for
energy production under pathological conditions, including ketone bodies, as was
exhibited in relevant experiments in both murine and human myocardial
tissue; in the former, increased expression of the Bdh1 gene
was observed, encoding the enzyme for the first step in oxidation of
A product of aerobic metabolism, reactive oxygen species (ROS) [68], are
commonly produced in cardiomyocytes by enzymes such as NADPH oxidases (NOX2,
NOX4), xanthine oxidoreductase, or nitric oxide synthase (NOS), and are usually
neutralized by antioxidant enzyme systems, including catalases, glutathione
peroxidases (GHSPx) and superoxide dismutases (SOD) [69]. Mechanical stress and
increased pressure seem to be a stimulus for increased activation of NOX2, 4 and
NOS (NOS decoupling) [70], with NOX2 also activated after myocardial infarction
(MI) [71]. ROS are also a by-product of the mitochondrial electron transport
chain (ETC), with a decrease in oxidative phosphorylation causing an increase in
ROS production [72]. One mechanism that may lead to increased ROS in this setting
could be increased Na
Additional ROS sequelae include inflammation, owing to activation of
NF-
Inflammation during cardiac remodelling is associated with the various stages of
atherosclerotic disease, from early atherogenesis, to plaque rupture and MI, thus
playing a role in the remodelling that occurs in ischemic heart disease (IHD)
[86]. Various molecules might become a nidus for an inflammatory response,
including cholesterol crystals, ROS, ischemia, as well as reperfusion injury
[87], promoting the formation of the NLRP3 inflammasome, which will activate
pathways within cells of the innate immune system, like resident macrophages and
monocytes [88]. Eventually, this will cause the production of IL-Ib and the
activation of TNF-
Epicardial fat has also emerged as a significant nidus for inflammation that may
contribute to pathological cardiac remodelling and as a result, dysfunction;
epicardial fat deposits possess unique secretomic characteristics, and exhibit
both thermogenic functions reminiscent of brown-fat adipose tissue, as well as
secrete factors that may be harmful to myocardial tissue, contributing to local
inflammation and fibrosis. These factors include pro-inflammatory
proteins, such as interleukin-1b (IL-1b), interleukin-6 (IL-6), tumor necrosis
factor (TNF-
Ca
Ca
In general, intracellular Na
Based on the ‘energy-starvation hypothesis’, pathological changes in metabolic
processes usually precede changes in contractile function during pathological
remodelling, eventually leading to hypertrophy and heart failure; preferential
use of ATP derived from glycolysis for NKA function might in part explain the
accumulation of Na
The reason for intracellular Na
Activation of the sympathetic, as well as renin-angiotensin-aldosterone (RAAS) systems appears to contribute to the overall progression of remodelling. The sympathetic system mediates its effect mainly through the release of noradrenaline (NA) [112], with relevant alterations including increased or decreased nerve density in the affected myocardium, as well as altered neurotransmitter production, which in turn may cause increased excitability [113]. Notably, the disruption of circadian rhythm could also facilitate cardiac remodelling and further increase levels of noradrenaline (NA) [114]. RAAS mediates its effects through an increase in aldosterone production [115], by binding to the AT1R receptor, affecting the vascular tone and blood volume, inducing fibrotic and hypertrophic changes, increasing ROS production and inflammation. MiRNAs may also play a role in this process, since they have been found to mediate some of these effects [116].
All these molecular and cellular alterations (summarized in Fig. 1), seem to have an effect on macroscopic heart dimensions, affecting parameters such as left ventricular wall thickness (LVWT), left ventricular cavity diameter in systole or diastole (LVDS, LVDD respectively), and associated volumes [117]. This in turn can also affect the ejection fraction (EF) as well as the general shape of the left ventricle and the physics of cardiac contraction [1, 37, 118].
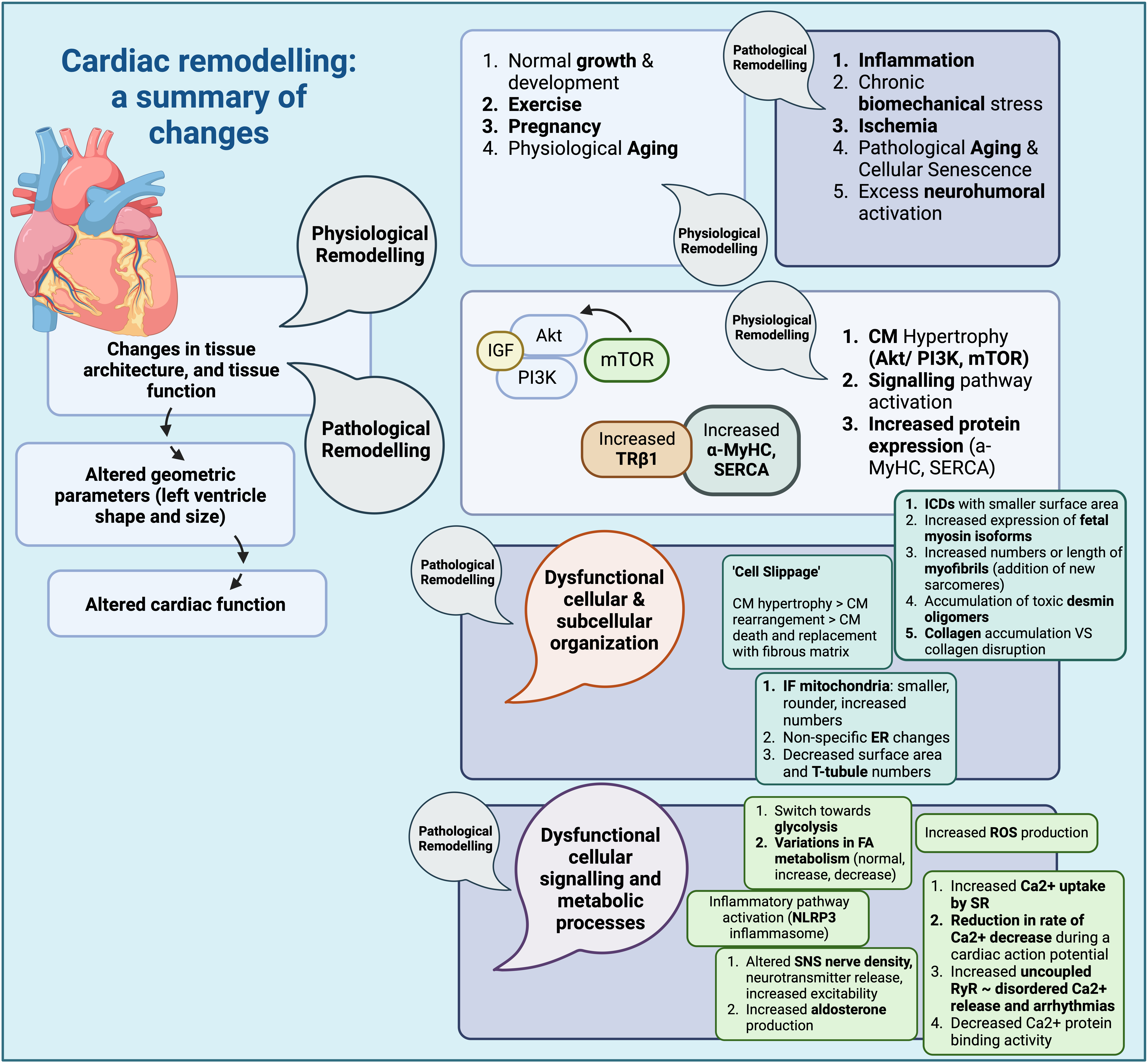 Fig. 1.
Fig. 1.Cardiac remodelling: a summary of changes (Created with
BioRender.com). Summary of structural changes and changes in signalling
pathways, as well as other cellular processes during cardiac remodelling. CM,
cardiomyocyte; IGF, insulin growth factor; PI3K, phosphoinositide 3 kinase; Akt,
protein kinase B; mTOR, mammalian target of rapamycin;
Glucose transporters within cardiac tissue comprise both glucose transporters
(GLUT) and sodium glucose cotransporters (SGLT) [119, 120, 121, 122, 123, 124, 125, 126]. GLUT1 and GLUT4 are
the most commonly expressed forms, in fetal and adult hearts, respectively [127].
While both mediate glucose uptake in cardiomyocytes, GLUT1 is often described as
insulin-independent, while GLUT4 is described as insulin-dependent [128, 129, 130, 131, 132].
Concerning SGLTs, studies that examine SGLT1 regulation within cardiac tissue
have pointed to a possible role for leptin, which seems to increase cardiac SGLT1
expression. Furthermore, SGLT1 has been shown to colocalize with GLUT1 receptors
and, at least in murine models, to increase with age. However, in terms of
function, some studies do indicate a role for cardiac SGLT1 receptors in glucose
uptake [132], while other differentiate between different cardiac SGLT1 variants,
with some having no role in glucose uptake, due to the absence of glucose and
Na
As expected, the expression and activity of glucose receptors could be altered
depending on the underlying states of the disease (summarized in Table 5 and Fig. 2), for example in the case of GLUT1, it appears to be overexpressed in chronic
hypoxic states, possibly through HIF-1a binding. Furthermore, relevant studies
have indicated a protective effect for increased GLUT1 expression in preventing
cardiomyocyte death under low O
| Receptor | Function |
| GLUT1 | Overexpression in chronic hypoxic states (HIF-1a)-prevents cardiomyocyte death under chronic states of low oxygen tension |
| Increased expression, in post-MI remodelling (rat models) | |
| Decreased expression, in late-stage heart failure (human samples) | |
| Overexpressed in pathological cardiac hypertrophy | |
| GLUT4 | Downregulated in pathological cardiac hypertrophy |
| Downregulated in diabetes (impedes glucose utilization by cardiomyocytes) (reversal with insulin administration of AT1R blockade) | |
| SGLT1 | Downregulation in murine models of diabetes |
| Overexpression in murine models of ischemia by coronary artery ligation | |
| Overexpression in human specimens of cardiomyopathy (diabetes, ischemia) | |
| Upregulation during cardiac tissue injury | |
| Role in glucose uptake contested with conflicting results from different studies |
GLUT1, glucose tansporter 1; SGLT, sodium glucose cotransporter; HIF-1a, Hypoxia inducible factor-1a; MI, myocardial infarction; AT1R, angiotensin 1 receptor.
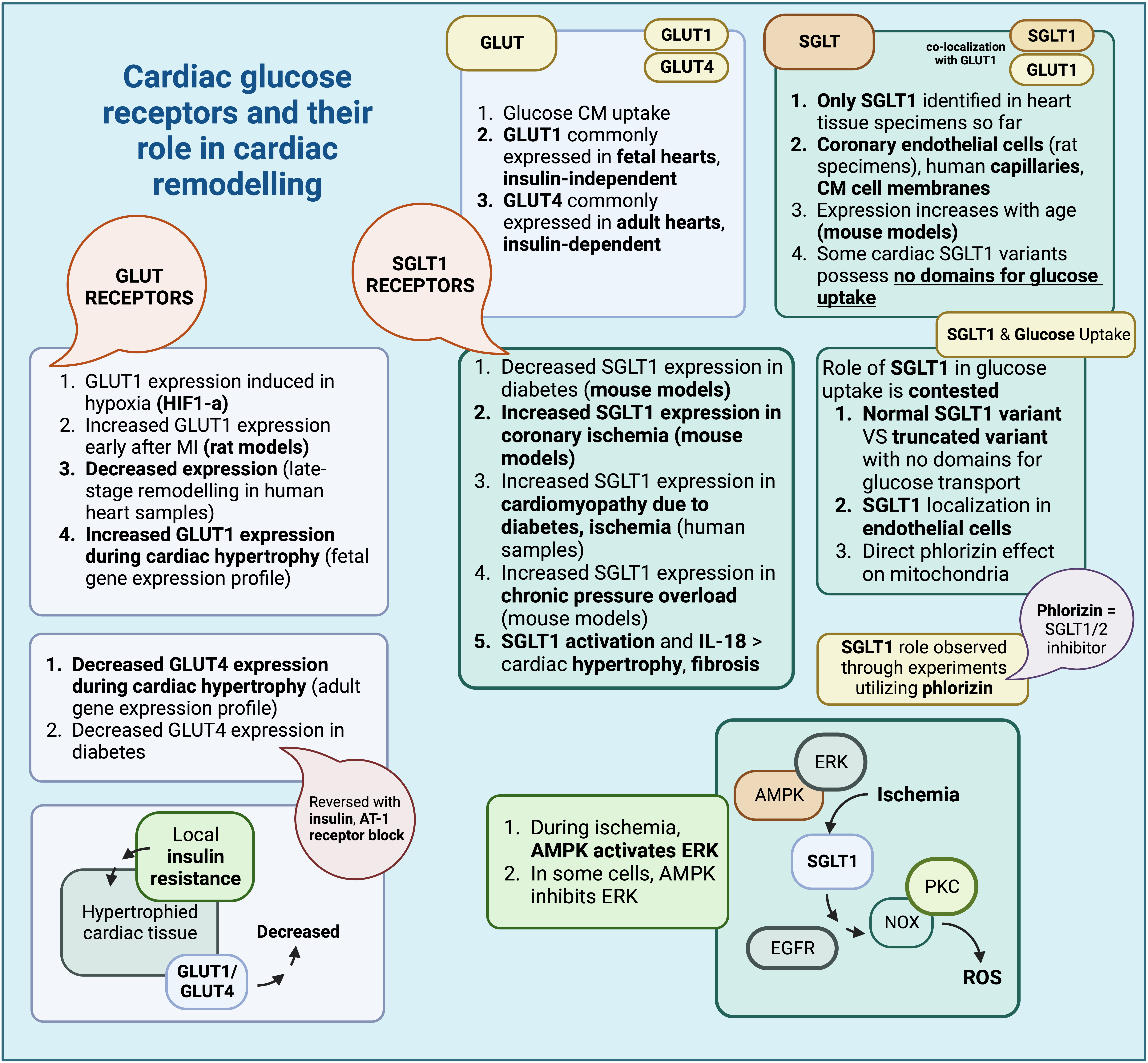 Fig. 2.
Fig. 2.Cardiac glucose receptors and their role in cardiac remodelling (Created with BioRender.com). Summary of glucose receptor characteristics found within cardiac tissue and their respective roles in cardiac remodelling. GLUT, glucose transporter; SGLT, sodium glucose co-transporter; HIF-1a, hypoxia inducible factor-1 a; AT-1, angiotensin-1; AMPK, adenosine monophosphate-activated protein kinase; ERK, extracellular signal regulated kinase; CM, cardiomyocyte; IL-18, interleukin-18; PKC, protein kinase C; NOX, NADPH oxidase; EGFR, epidermal growth factor receptor; ROS, reactive oxygen species.
In the case of SGLT receptors, recent evidence appears to point to a role for them in the pathogenesis of cardiac remodelling; this is true for SGLT1 receptors, the only SGLT receptor type found within cardiac tissue so far. Oftentimes, SGLT1 derangements are associated with metabolic disturbances; indeed, SGLT1 expression appears to decrease in murine models of diabetes but increases in murine models of ischemia through coronary artery ligation. In human specimens, SGLT1 seems to increase in cases of cardiomyopathy due to diabetes and ischemia. Therefore, some authors theorize that upregulation of cardiac SGLT1 is an adaptation to tissue injury [132]; in fact, administration of phlorizin, an inhibitor of SGLT1, after restoration of coronary blood flow, did not lead to any restoration of ventricular function. ATP seems to be depleted earlier in these cases as well; thus, SGLT1, as observed in these experiments, might play a role in glucose uptake during injury [138]. However, conflicting reports do exist, which attribute no significant role for cardiac SGLT1s in glucose uptake whatsoever [133]; these different results could be perhaps attributed to various factors, including the presence of a normal and a truncated variant of SGLT1 [133], localization of SGLT1 receptors in endothelial cells [130], as well as the direct effect of phlorizin in mitochondria and mitochondrial ATPase [139].
Additional experiments evaluating the role of SGLT1 during ischemia and ischemia-reperfusion indicated that SGLT1 might be responsible for the observed myocardial injury, since SGLT1 knockdown was associated with milder issue injury. SGLT1 expression during ischemia is indirectly upregulated by adenosine monophosphate-activated protein kinase (AMPK) through extracellular signal regulated kinase (ERK) [140], although in other cells, AMPK appears to inhibit ERK instead [141]. SGLT1 causes PKC and NOX activation through EGFR, eventually contributing to oxidative stress and ROS species production [140]. There also appear to be conflicting results with respect to the association between cardiac injury and experimental SGLT1 inhibition, depending on whether the inhibition is carried out pharmacologically or through RNAi, with the resulting injury being exacerbated or ameliorated, respectively [140]. SGLT1 expression also appears to be upregulated in cases of chronic pressure overload, as shown in relevant murine models of transverse aortic constriction (TAC), and, along with IL-18, it might have a role in the development of subsequent cardiac hypertrophy and fibrosis [142]. In fact, transgenic expression of SGLT1 is associated with higher rates of tissue hypertrophy and cardiomyocyte size, along with signs of ventricular dysfunction ameliorated with transgenic SGLT1 suppression [143].
With the emerging role of SGLT1 receptors during ischemic myocardial injury [140], their role in cardiomyocyte hypertrophy after injury [142], as well as results of preclinical studies of SGLT1 [138, 140], it seems that SGLT1 inhibition might be beneficial to the injury and possible remodelling observed after myocardial ischemia. Phlorizin, a non-selective SGLT inhibitor, has been used in preclinical studies for experimental inhibition of SGLT1 [138, 144], and although it can correct hyperglycemia, it has poor oral bioavailability. Its relevant analogues, including glucoside O and C, have been used to derive compounds such as empagliflozin and canagliflozin, approved drugs for the treatment of diabetes mellitus [145, 146].
SGLT1 inhibition can be achieved through selective SGLT1 inhibitors, or through
substances that affect both SGLT1/SGLT2 receptors [145]. Some examples of SGLT1
inhibitors include KGA-2727, with promising results in mitigating ventricular
remodelling and preventing increases in cardiomyocyte size, ANP, BNP, and IL-18,
in murine models of myocardial infarction [147]. Mizagliflozin, which has
completed phase II of clinical trials [145], appears to be useful in mitigating
cardiomyocyte apoptosis, as well as the resulting cardiomyopathy observed in
diabetes, through inhibition of the Jun N-terminal Kinase (JNK) and p38 pathways [148]. Sotagliflozin, a
dual SGLT1/SGLT2 inhibitor, seems to alleviate some of the myocardial injury
observed in pressure overload models, although this has been observed only in
subjects with a normal diet [149]. Sotagliflozin is currently being used in
Europe, approved for patients with type 1 diabetes and a body mass index (BMI) greater than 27
kg/m
Relevant cardioprotective mechanisms induced by SGLT1 inhibition, summarised in Fig. 3, could include down-regulation of the JNK and p38 MAPK pathways, as well as prevention of cardiac fibrosis, which is also involved in the pathogenesis of diabetic cardiomyopathy (DCM), a diabetes-associated cardiomyopathy associated with increased cardiomyocyte size, cardiomyocyte apoptosis, and myocardial fibrosis, leading to ventricular dysfunction [148, 150, 151]. There are studies that point to a possible detrimental effect of diabetes on ventricular function and its contribution to pathological remodelling [152, 153], something that could be ameliorated with SGLT1 inhibition [145]. Another mechanism of action might include inhibition of the EGFR-ERK-PKC-Nox pathway (KGA-2727), particularly in the injured myocardium after myocardial infarction, affecting the subsequent remodelling [140, 147]. Finally, inhibition of SGLT1 might also prevent angiotensin II-mediated activation of SGLT1 receptors in vascular endothelial cells, in turn preventing ROS accumulation through NADPH activation, decreasing the incidence of endothelial senescence [154].
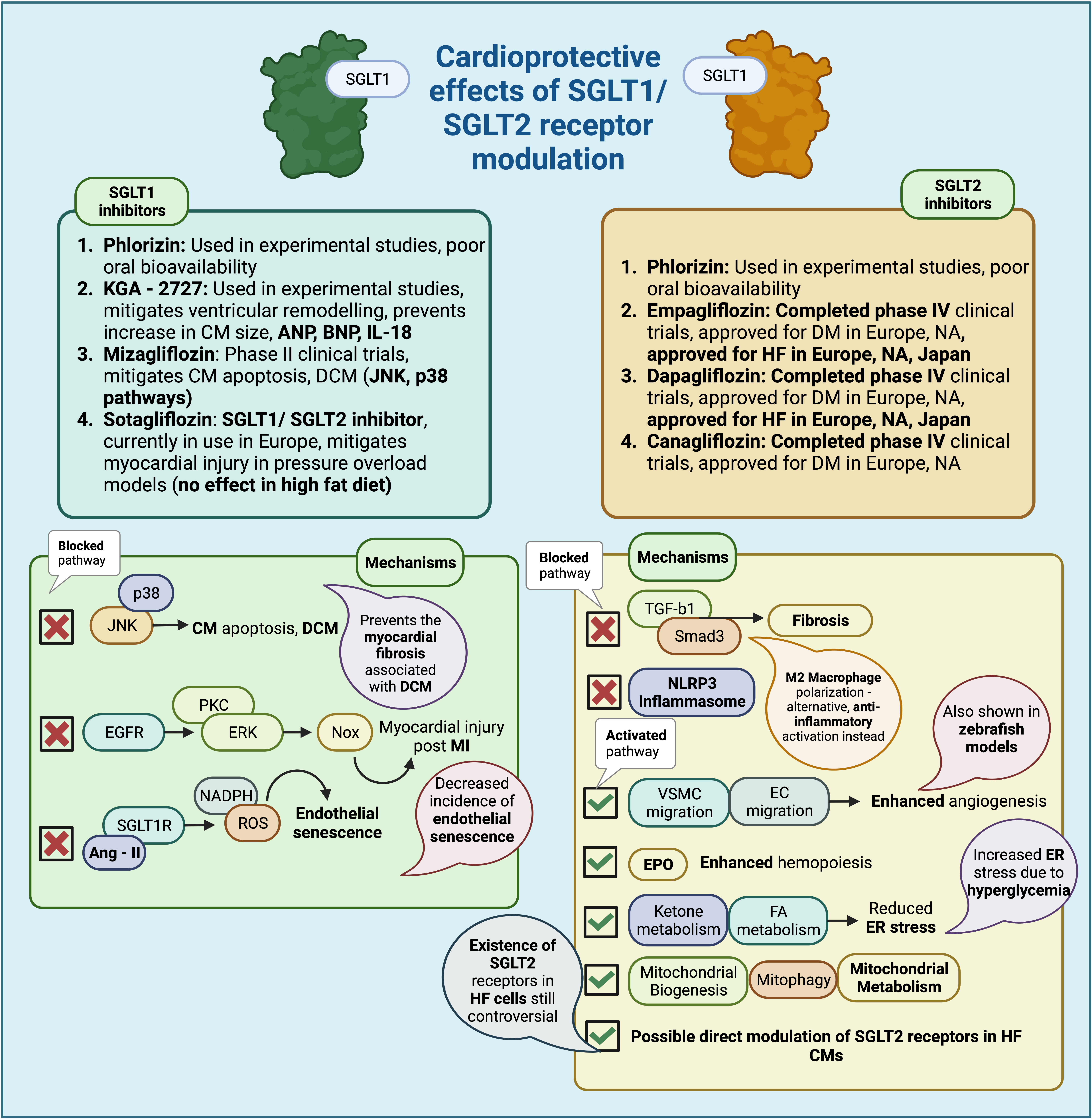 Fig. 3.
Fig. 3.Cardioprotective effects of SGLT1/SGLT2 receptor modulation
(Created with BioRender.com). Summary of the cardioprotective effects of SGLT1
and SGLT2 receptor inhibition, via inhibition or facilitation of specific
biological pathways. SGLT, sodium glucose co-transporter; CM, cardiomyocyte; ANP,
atrial natriuretic peptide; BNP, brain natriuteric peptide; IL-18, interleukin
18; DCM, diabetic cardiomyopathy; JNK, Jun N-terminal kinase; HF, heart failure;
EGFR, epidermal growth factor receptor; PKC, protein kinase C; ERK, extracellular
signal related kinase; NOX, NADPH oxidase; MI, myocardial infarction; Ang-II,
angiotensin II; NADPH, Nicotinamide Adenine Dinucleotide Phosphate Hydrogen; ROS,
reactive oxygen species; HF, heart failure; FA, fatty acid; ER, endoplasmic
reticulum; TGF-
Although no SGLT2 receptors have been consistently found within cardiac tissue in relevant experiments, SGLT2 inhibition might also have cardioprotective effects through different mechanisms. The associated lack of hypoglycemia with SGLT2is, which could potentially exacerbate cardiac injury through increased sympathetic activation, also presents a favorable profile for use in cases of cardiovascular remodelling [37, 155]. Examples of SGLT2is include canagliflozin, empagliflozin, and dapagliflozin, all approved for the treatment of diabetes in both Europe and North America (NA); moreover, dapagliflozin and empagliflozin have also been approved for the treatment of HF and kidney disease, in Europe, NA and Japan [145].
Cardioprotective mechanisms of action for SGLT2is include weight loss, possibly related to inhibition of sodium and glucose reabsorption, reduction of plasma insulin and glucose levels, reduction in blood pressure, due to reduced sodium reabsorption and, over time, reduced RAAS activation, as well as increased erythropoietin (EPO) production which improves erythropoiesis, thus aiding in better tissue oxygenation [156]. Additional effects, more closely related to cardiovascular remodelling, include positive effects on inflammation, fibrosis-related signaling pathways, angiogenesis, cellular organelle function, and direct cardiomyocyte effects [157]. Mechanisms more closely related to the potential of SGLT2is to ameliorate or reverse the effects of cardiovascular remodelling include effects on inflammation, signalling pathways involved in cardiac fibrosis and inflammation, subcellular organelle function and ketone metabolism, angiogenesis, and perhaps possible direct effects on cardiomyocytes [156].
Anti-inflammatory effects of SGLT2is include inhibition of the NLRP3
inflammasome, through reduction of intracellular Ca
SGLT2is also enhance mitochondrial metabolism [160], facilitate the generation of new mitochondria (mitochondrial biogenesis) [164], and removal of senescent mitochondria (mitophagy) [165]. endoplasmic reticulum (ER) stress, often induced by high glucose concentrations, can also be alleviated by SGLT2is, through induction of enzymes involved in ketone and FA metabolism [166]. SGLT2is may also act upon and modulate ketone metabolism, an important aspect of their cardioprotective properties; it seems that SGLT2is can mildly increase levels of ketone compounds, levels of genes involved in ketolysis, as well as associated uptake of ketones, fatty-acids (FA) and branched chain amino acids (BCAA) [167]. This increase in ketone and FA uptake has been observed to occur in relevant swine intravascular balloon occlusion models, ameliorating the reduction in FA uptake occurring after the initial insult [58, 168].
SGLT2is have been further shown to act on pathways involved in angiogenesis, which, when disturbed, could contribute to adverse cardiac remodelling, especially after MI [169]. Pharmaceutical agents, such as sotagliflozin, have been used to enhance migration of vascular endothelial cells and smooth muscle cells [170], while others, such as empagliflozin and dapagliflozin, have been shown to enhance angiogenesis in an embryonic zebrafish model [171]. SGLT2is also affect hemopoiesis, by up-regulating EPO secretion by the kidney [5, 172].
Finally, SGLT2is may exert direct effects on cardiomyocytes, perhaps through the
presence of SGLT2 receptors on cardiomyocytes themselves; more specifically, one
relevant study detected the presence of SGLT2 receptors in end-stage HF
cardiomyocytes, as well as transplanted cardiomyocytes within diabetic hosts,
with overexpression of the SGLT2 receptor associated with cardiomyocyte metabolic
derangements [173]. In addition, SGLT2is have recently been shown to affect ion
transporters within cardiomyocytes, including Ca
Stem cells are undifferentiated cells, with the ability for self-renewal and generation of differentiated progeny. There are many stem cell types, with variable potential for progeny generation, including mesenchymal stem cells (MSC), i.e., multipotent stem cells capable of generating tissues such as bone, muscle, and cartilage, and cardiac stem cells (CSC) [178, 179]. On the other hand, progenitor cells, are more lineage-specific and usually generate one or only a few lineage-specific cell types [180].
Local stem and progenitor cells have been associated with a number of pathological remodelling processes (Table 6, Ref. [181]), including the progressive change in cardiac tissue composition that is related to aging (mainly attributed to senescence of local progenitors [13]), and the contribution of local cardiac MSCs [182] to the pathological fibrosis observed during cardiac remodelling [12]. Some of these progenitors, including endothelial progenitors (EPC), are particularly important during cardiac tissue injury, as they contribute to local blood vessel generation, either de novo (vasculogenesis), or through existing vessels (angiogenesis) [183, 184]. Furthermore, with their secretomic profile, they contribute to multiple repair and regeneration processes [183], allow homing of other stem/progenitor cells, including cardiac progenitors [185], prevent MSC-derived deleterious effects such as fibrosis [186], as well as contribute to mitigation of inflammation and oxidative stress [187].
| Cell type | Alteration | Effects/Additional information |
| MSC | Pro-inflammatory, pro-fibrotic phenotype change | PDGFR |
| HSPC | Reduction in circulating HSPC populations | ‘Three-Department’ model; defective mobilization from the bone marrow (BM)/damage to the niche itself, decreased survival of circulating HSPC, impaired trafficking of pro-vascular progenitors to target sites. |
| Increased HSPC retention in the bone marrow, increased HSPC differentiation towards myeloid lineages. | ||
| EPC | Reduction in EPC populations, EPC senescence | Continuous cardiovascular injury and EPC mobilization causing EPC depletion, increased oxidative stress, EPC senescence (increased SA |
| Reduction in EPC levels (CD34+ KDR+EPC) increases risk of adverse cardiovascular events and restenosis after cardiovascular intervention. | ||
| CSC | CSC senescence | CSC senescence (p16, SA |
| Elimination of senescent cells in mouse models improved cardiac function and remodelling after myocardial injury (Salerno et al., 2022 [181]). |
PDGFR
Although MSCs have been known to contribute to tissue repair and mitigation of
inflammatory responses [188, 189], some endogenous MSCs have been shown to exhibit
a pro-inflammatory and pro-fibrotic phenotype, possibly contributing to the
fibrotic response during pathological cardiac remodelling; this seems to occur
either through upregulation of the of the PDGFR
Progenitor cells can be affected during pathological remodelling processes, usually due to chronic dysglycemia and diabetes; haematopoietic stem and progenitor cells (HSPC), for example, which can also contribute to the provascular cell pool [190], and more specifically, CD34+ HSPCs, have been found in reduced numbers in the peripheral circulation, in both T1DM and T2DM [191, 192]. The ‘three-department’ model has been developed to explain this defect in peripherally circulating HSPCs; defective mobilization from the bone marrow (BM) (stem cell niche) as well as damage to the niche itself, decreased survival of circulating HSPC in the periphery (something not yet proven in human specimens), and impaired trafficking of pro-vascular progenitors to target sites, are all believed to contribute to the apparent decrease in pro-vascular HSPC populations in diabetes. Furthermore, a shift in differentiation balance toward myeloid progenitors also appears to occur, possibly due to increased production of S100A8/9 proteins (alarmins) by pro- inflammatory cells. In the bone marrow, this leads to the release of oncostatin M (OSM) and CXCL12 production, which increases retention of HSPC, and promotes their differentiation toward a myeloid lineage, while in the periphery, this mainly promotes inflammation in target organs [193].
Endothelial progenitor cells (EPCs), derived from different sources, including
the vascular wall [194], have a positive effect on post-injury cardiac
remodelling, by aiding in myocardial repair [183]; decreased numbers of EPCs due
to a number of factors, including continuous cardiovascular injury and
mobilisation of EPCs with subsequent depletion in numbers, increased oxidative
stress, and even EPC senescence (characterized by increased SA
Although the contribution of EPCs to the mitigation of injury has been shown, defining an EPC population has been a point of debate between different studies, since markers such as VEGFR-2 and CD34 can also be expressed by myeloid cell lineages; relevant studies differentiate between true EPCs, positive for CD34/VEGFR-2, and negative for CD45/CD14, that can generate both endothelial cells (EC) in vitro and integrate in growing vessels in vivo, and circulating angiogenic cells (CAC), expressing both endothelial (CD34/VEGFR-2) and hemopoietic lineage markers (CD45/CD14), and while capable of in vitro angiogenesis, they cannot integrate within growing vessels in vivo [198, 199]. Aldehyde dehydrogenase (ALDH) has been recently used to differentiate vascular progenitors from other hematopoietic stem and progenitor cell populations, with pro-vascular cells exhibiting high ALDH activity; this, along with other endothelial-specific markers, has been used to better identify vascular progenitor populations [198].
Finally, cardiac stem cell (CSC) senescence has also been shown to contribute to
the development of varying disease phenotypes. A wide range of cardiac stem cell
(CSC) populations have been identified, some existing in adult hearts and others
in embryonic hearts [200]; CSC senescence, denoted by an increase in markers such
as p16, SA
Recent research on the mechanisms with which stem and progenitor cells might contribute to the progression of pathological remodelling gave way to studies examining the effect of manipulation of the SGLT receptor on stem and progenitor cells in this context [12, 196, 202]. The need to answer this particular research question might be further explained by the effect of diabetes and dysglycemia on progenitors, as well as the associated cardiovascular risk [193], and the pathological remodelling that may occur in cases of diabetes [150]. Although it would be logical to assume that some effect on progenitor populations would be expected, given the beneficial effect of SGLTis on remodelling, so far studies have produced conflicting results; the primary setting for this proposed effect has mostly been investigated in the setting of diabetes mellitus, as SGLTis have been used primarily for their cardioprotective and antidiabetic effect.
SGLT inhibition and its effect on stem and progenitor cells in human patients was investigated for the first time in 2018, with a randomized clinical trial (RCT), by Bonora et al. [203]; dapagliflozin was evaluated in a total of 33 patients with T2DM for 12 weeks, although an extension period was added, up to 74 weeks [203]. Additionally, another 15 patients were added, which in this case received empagliflozin. The type of cells evaluated were circulating progenitors expressing CD34 antigen (CD3+-CPC) and endothelial progenitors, identified through their additional expression of KDR, a marker found primarily in cells of endothelial lineage (CD34+ KDR+-EPC). There was no direct significant effect on these cell populations in response to SGLT2i treatment, although there was some increase in progenitors after an extended period, which was attributed to better glycemic control by the researchers, correlated with HbA1c levels, rather than the SGLT2i treatment itself [203].
Another similar substudy followed in 2019, part of the EMPA-HEART Cardiolink-6
trial [204], by Hess et al. [205], in which researchers evaluated the
role of empagliflozin in circulating vascular progenitors. Vascular progenitors
were defined as a heterogeneous cellular group that includes bone marrow-derived
EPCs (BM) (BM-EPC), angiogenic HSPCs, as well as monocyte-derived angiogenic
macrophages [205]. Their levels were evaluated in patients, both at baseline and
within 6 months after empagliflozin treatment. An additional marker was used to
identify vascular progenitors, aldehyde dehydrogenase (ALDH), a detoxification
enzyme found primarily within immature progenitors [206]. ALDH has been shown to
better aid in identifying such cells, due to less variability in the expression
of the ADLH gene, in contrast to other markers, such as CD133 and CD34,
whose expression could vary depending on the stage of development [206]. Another
cellular aspect that was quantified to better aid in cellular characterization
was intracellular granular complexity, also termed side scatter property (SSC);
thus, in this study, EPCs were defined, not only based on CD133 and CD34
expression, but also on SSC levels and ALDH expression [205]. High and middle
complexity was used to denote pro-inflammatory granulocytes and monocytes, while
low SSC was used to identify a heterogenous group of vascular progenitors with
high expression of ALDH [190, 205]. Hence, vascular progenitors in this study were
defined as a heterogeneous cellular group of ALDH
Some of the results of this study [205] seem to contradict some of the results
of a previous study by Bonora et al. [203], while in the long term, even
in the latter, cells staining positive for CD34 and VEGFRR2 also appeared to
increase [203], even though a different SGLT2i was used each time. It seems that
more detailed characterisation of vascular progenitor cells generated a different
result in the study by Hess et al. [205]; the criteria used to
characterise potential vascular progenitors generated further disagreement
amongst research groups, since some authors noted that the populations identified
through ALDH expression were heterogeneous, containing both vascular and
hemopoietic progenitors [207]. However, the angiogenic potential of
ALDH
Additional studies followed, examining the role of SGLT2 inhibitors, such as dapagliflozin, in the mobilization of hematopoietic cells from the bone marrow (BM); this was examined in mice models of T1DM and T2DM, as well as mouse models of carotid endothelial injury. While an increased ratio of granulocytes/lymphocytes was observed in subjects with diabetes, possibly due to the enhanced myelopoiesis associated with hyperglycemia, dapagliflozin did not appear to have any effect. An interesting finding in this study is the observation of CD49d+ granulocyte mobilization after dapagliflozin administration, which helped vascular repair within 3 days, in a murine model [210]. The role of SGLT2 inhibitors, and more specifically empagliflozin, on vascular progenitor populations was again, more recently examined in a substudy by Bakbak et al., 2023 [211], included within the EMPA-HEART 2 Cardiolink-7 trial [212]; in this case however, this effect was studied irrespective of T1DM or T2DM status, as only patients with cardiovascular risk factors were included. After 6 months of treatment, angiogenic progenitor cells as well as regenerative monocyte progenitor cells were found to be increased, while pro-inflammatory progenitors were found to be decreased. Thus, it seems that SGLT2 inhibitors can indeed exert a cardioprotective effect, through increasing angiogenic progenitors, even in the absence of diabetic status [211].
Finally, the administration of dapagliflozin also seemed to improve hematopoietic cell mobilization, as well as aid in the restoration of the vascular endothelium in areas of tissue damage, in relevant models of carotid injury; a mechanism for this was shown to be induced through the recruitment of BM-derived cells, through experimental inhibition of CXCR4 with the compound AMD3100, co-administered with dapagliflozin. Since SGLT2 expression has been detected at very low levels in the bone marrow, the authors concluded that a direct effect on the bone marrow by dapagliflozin seemed unlikely [210]. This mechanism could counteract the observed depletion of circulating angiogenic HSPCs in the diabetic state; furthermore, modulation of pro-inflammatory cell production from existing HSPC populations appears to mitigate vascular dysfunction, as well as endothelial damage [213].
In essence, the association between SGLT modulation and progenitor cells is relatively newfound, and therefore contradictory studies still exist (summarized in Table 7, Ref. [203, 204, 205, 208, 210, 211, 213]); if solidified by more studies, it could serve as additional clarification on the positive effects of SGLT2 inhibitors on the cardiovascular system [14].
| Publication | SGLTi | Cell | Observations |
| Bonora et al., 2018 [203] | Dapagliflozin, Empagliflozin (on additional 15 patients) | CD34+-CPC | No direct significant effect in response to SGLT2i treatment. |
| CD34+ KDR+-EPC | Modest increase in progenitor numbers after an extended period, attributed to better glycemic control. | ||
| Verma et al., 2019 [204] | Empagliflozin | ALDH |
Substudy of the EMPA-HEART Cardiolink-6 trial (Verma et al., 2019 [204]). |
| Hess et al., 2019 [205] | Novel method to identify vascular progenitors, ALDH and SSC (intracellular complexity)-vascular progenitors from a variety of populations (BM-EPCs, angiogenic HSPCs, angiogenic monocytic-derived macrophages). | ||
| Bakbak et al., 2023 [211] | Empagliflozin | ALDH |
Substudy of the EMPA-HEART Cardiolink-7 trial (Connelly et al., 2023). |
| Evaluation of 3 progenitor cell populations after 6 months of empagliflozin treatment vs placebo; angiogenic progenitors and regenerative monocyte progenitors were increased, while pro-inflammatory progenitors were decreased. | |||
| No T1DM, T2DM associations in this study-only patients with cardiovascular risk factors were included. | |||
| Cooper et al., 2018 [208] | N/A | ALDH |
ALDH |
| Albiero et al., 2021 [210] | Dapagliflozin | CD49d+ granulocytes | T1DM, T2DM murine models, murine models of endothelial injury. |
| Dapagliflozin had no effect on the increased granulocyte/lymphocyte ratio observed in diabetic states. | |||
| Mobilization of CD49d+ granulocytes, after dapagliflozin administration, facilitating vascular repair within 3 days. | |||
| Hess et al., 2021 [213] | Dapagliflozin | HSPCs | Improved HSPC mobilization, recruitment of BM-derived cells, modulation of pro-inflammatory cell production from existing HSPC populations tackles vascular dysfunction and endothelial damage in relevant models. |
CPC, cardiac progenitor cells; KDR, kinase insert domain receptor; EPC, endothelial progenitor cells; SGLT2i, sodium glucose co-transporter 2 inhibitor; ALDH, aldehyde dehydrogenase; SSC, side scatter property; BM-EPC, Bone marrow endothelial progenitor cells; HSPC, hematopoietic stem and progenitor cells; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; BM, bone marrow.
Cardiac remodelling is a complex process that affects multiple subcellular structures and processes, from mitochondria and cell membranes, to gene expression and protein composition. Eventually, changes in tissue micro- and macroarchitecture can occur, impacting ventricular function and cardiac output. The mechanisms involved in cardiac remodelling have been extensively researched with new evidence constantly being discovered. Amongst the various pathophysiological mechanisms that can contribute to cardiac remodelling, various local stem and progenitors cell types seem to play a role. Cardiac MSCs contribute to the fibrotic reaction, while decreased populations of vascular progenitors and HSPCs, as well as senescence of local cardiac stem cells can alter the healing of local tissue injury and further contribute to the exacerbation of post-injury remodelling. Stem and progenitor cell populations have also been found to be affected during dysglycemic states and diabetes.
Glucose receptors, such as GLUT and SGLT, have been found at various locations, and though GLUT1 and GLUT4 exist within cardiac tissue, evidence pointing to the existence of cardiac SGLT1 and possibly SGLT2, is not yet well solidified. Notably, it has been proposed that the different locations of SGLT2 and SGLT1 could be related to different efficacy and safety characteristics between dual (i.e., sotagliflozin) and selective for SGLT2 inhibitors (i.e., empagliflozin) [214, 215, 216].
The association between SGLT inhibition and stem/progenitor cells has only recently emerged, with existing studies still producing conflicting results. Although vascular progenitors seem to be affected by SGLT2 modulation in some studies, others do not establish a significant connection. One of the reasons for these contradictory results seems to be the criteria used to identify endothelial progenitor populations, since different studies use different criteria. Therefore, more studies must be conducted so that universal criteria for defining each stem/progenitor cell population are defined and accepted, to better help interpret test results from different studies. Existing studies have explored the association between endothelial progenitors and SGLT2 modulation in the context of diabetes mellitus, as well as in patients with cardiovascular risk factors, without diabetic status; it might be interesting to further examine the effect of SGLT2 modulation on vascular progenitors in other disease entities that may lead to pathological cardiac remodelling as well, to further establish this connection between SGLT2 modulation and progenitor cells. Finally, EPCs are not the only types of progenitor cell involved in pathological remodelling; additional studies might be warranted that evaluate the effects of SGLT2is, if any, on dysfunctional cardiac MSCs, and whether the resulting fibrosis could be mitigated.
The cardioprotective effects of SGLT2 inhibitors are evident, with positive effects observed in people with or without diabetes; in addition, derangements in stem and progenitor cell numbers or function have been shown to contribute to the progression of pathological cardiac remodelling. Although the association between SGLT2is and progenitors is relatively new, if solidified, it could facilitate better management of pathological aspects of cardiac remodelling, further mitigating cardiac dysfunction.
SLC5A1, solute carrier family 5 member 1; SLC5A2, solute carrier family 5 member
2; PCT, proximal convoluted tubule; SGLT2, sodium glucose co-transporter 2;
SGLT1, sodium glucose co-transporter 1; MSC, mesenchymal stem cells; CSC, cardiac
stem cells; EPCs, endothelial progenitor cells; HSPCs, hemopoietic stem
progenitor cells; HF, heart failure; HFA, heart failure association; T1DM, Type 1
Diabetes Mellitus; T2DM, Type 2 Diabetes Mellitus; DM, diabetes mellitus; DCM,
diabetic cardiomyopathy; HOCM, hypertrophic cardiomyopathy; LV, left ventricle;
CO, cardiac output; BCAA, Branched chain amino acid; DHP, Dihydropyridine
receptor; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3 kinase;
Rac1, Ras-related C3 botulinum toxin subtrate 1; Akt, protein kinase B; CM,
cardiomyocyte; IGF, insulin-like growth factor; SIRT6, sirtuin 6; SR,
sarcoendoplasmic reticulum;
TMS and KCC made substantial contributions to conception and design of the manuscript, carried out the literature search, drafted and wrote the manuscript. TK assisted in literature research, DM, FM, CDM and DK reviewed the literature and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Figures were created with BioRender.com.
This research received no external funding.
TK has received honoraria for lectures from AstraZeneca, Boehringer Ingelheim, Pharmaserve Lilly, and Novo Nordisk, for advisory boards from Novo Nordisk and Boehringer Ingelheim, and has participated in sponsored studies by Eli-Lilly and Novo Nordisk. C. David Mazer reports advisory board honoraria and/or consulting fees from Amgen, AstraZeneca, BioAge, Boehringer Ingelheim, Cardior, PhaseBio and Sandoz; and reports stipends for serving on the Data and Safety Monitoring Board of Beth Israel Deaconess Medical Center, Cerus, and Takeda; Dr. Mazer is supported by a merit award from the University of Toronto Department of Anesthesiology, and holds the Cara Phelan Chair in Critical Care at St. Michael’s Hospital. TMS, KCC, FM, DM, DK have no conflicts of interest to report.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.