


1 Institute of Basic Medicine, North Sichuan Medical College, 637000 Nanchong, Sichuan, China
2 Department of Laboratory Medicine, Xichong County People’s Hospital, 637000 Nanchong, Sichuan, China
Abstract
The roles of small extracellular vesicles (sEVs) and mRNA modifications in regulating hepatitis B virus (HBV) transmission, replication, and related disease progression have received considerable attention. However, the mechanisms through which methyltransferase-like 3 (METTL3) and insulin-like growth factor 2 (IGF2BP2), key genes that mediate m6A modifications, regulate HBV replication in sEVs remain poorly understood. Therefore, this study investigated the molecular mechanisms through which the key molecules (METTL3 and IGF2BP2) in sEVs mediate m6A epigenetic modification to regulate HBV replication.
Small extracellular vesicles were extracted from the supernatants of HepG2.2.15 and HepG2 cells via ultracentrifugation, followed by purification with hepatitis B virus surface antigen (HepBsAg) immunomagnetic beads. The sEVs were characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS), and Western blotting (WB). Methylation enrichment in the two types of sEVs was analyzed by dot blotting and quantitative reverse transcription-PCR (RT-qPCR). The cells were treated with HepG2.2.15–sEVs transfected with either the METTL3 plasmid, METTL3 siRNA, the IGF2BP2 plasmid, or the IGF2BP2 siRNA. After 48 h, the expression of METTL3, IGF2BP2, and HBV DNA expressions were assessed via dot blotting, quantitative-PCR (qPCR), RT-qPCR, and WB. Co-immunoprecipitation (co-IP) was performed to investigate the interactions between METTL3 and IGF2BP2.
By conducting TEM, DLS, and WB analyses, we confirmed that the isolated sEVs exhibited typical characteristics. HepG2.2.15-derived sEVs presented elevated levels of m6A modifications, with increased METTL3 and IGF2BP2 mRNA and protein expression levels, respectively (p < 0.05). In the overexpression (OE)-METTL3 group, the expression levels of HBV pregenomic RNA (HBV pgRNA), HBV DNA, HBV relaxed circular DNA (HBV rcDNA), HBV covalently closed circular DNA (HBV cccDNA), HBsAg, hepatitis B virus core antigen (HBcAg), and hepatitis B virus e antigen (HBeAg) were significantly elevated compared to those in the control group (p < 0.01). In contrast, results for the small interfering (SI)-METTL3 group were the opposite. Similarly, in the OE-IGF2BP2 group, HBV pgRNA, HBV DNA, HBV rcDNA, HBV cccDNA, HBsAg, HBcAg, and HBeAg expression were greater than in the control group (p < 0.05), whereas the opposite results were recorded in the SI-IGF2BP2 group. Co-immunoprecipitation confirmed that METTL3 and IGF2BP2 interact synergistically.
Small extracellular vesicles increase METTL3 and IGF2BP2 expression, synergistically promoting HBV replication by regulating m6A modification levels.
Keywords
- HBV
- small extracellular vesicles
- N6-methyladenosine modification
- METTL3
- IGF2BP2
Hepatitis B virus (HBV) possesses a 3.2 kb partial double-stranded relaxed circular DNA (rcDNA) genome, which can be repaired in the nucleus to covalently closed circular DNA (cccDNA) [1, 2, 3]. The cccDNA can be transcribed to generate new pregenomic RNA (pgRNA), which can produce new rcDNA via reverse transcription. This rcDNA can be secreted into the extracellular environment via small extracellular vesicles (sEVs), thus facilitating the infection of additional hepatocytes [4, 5, 6]. The primary factors contributing to persistent HBV infection include the chromosomal integration of cccDNA into host cells and infection mediated by sEVs [7]. However, the precise regulatory mechanisms underlying these processes remain poorly understood.
Small extracellular vesicles (sEVs) are bilayer vesicle structures characterized by a cup-shaped morphology, with particle sizes between 30 and 200 nm [8, 9]. They are crucial in facilitating the transmission of HBV, disease progression, and the development of targeted therapy [10, 11, 12]. The study has indicated that interfering with extracellular vesicles significantly reduces HBV replication [13]. Moreover, engineered sEVs containing clustered regularly interspaced short palindromic repeats (Cas9/gR) and vesicular stomatitis virus-glycoprotein (VSV-G) can decrease HBV replication and cccDNA levels [14]. These findings highlight the close association between sEVs and the loading and dissemination of HBV viral particles, as sEVs contain various components, including HBV DNA, HBV RNA, and proteins [15, 16].
Studies have shown that the m6A sites in the hepatitis C virus (HCV) can
regulate the maturation of the virus in complex and undetermined ways [17, 18].
Some researchers found that N6-methyladenosine binding protein (YTHDC2)
mediates m6A modifications to regulate HCV internal ribosome entry site
(IRES)-dependent translation [19]. The conserved m6A motifs located in the
Study has found that the proteins methyltransferase-like 3 (METTL3) and insulin-like growth factor 2 (IGF2BP2) that act as a writer and a reader, respectively, function as coregulators of m6A modification during HBV infection, with METTL3 facilitating m6A enrichment in an IGF2BP2-dependent manner [28]. However, the precise mechanism by which sEVs mediate m6A epigenetic modifications to regulate HBV replication during viral packaging into sEVs for transmission to uninfected cells remains to be elucidated.
In our previous study, we found that METTL3 synergizes with IGF2BP2 to regulate HBV replication at the cellular level [29]. To further elucidate the specific molecular mechanisms through which sEVs mediate m6A modification and promote HBV replication via METTL3-IGF2BP2 synergy, we conducted this study. Unlike previous studies that focused on the cellular level, we regulated m6A modifications at the microscopic level in sEVs. We hypothesized that sEVs enhance HBV replication by modulating m6A levels through interactions between METTL3 and IGF2BP2. We examined changes in HBV replication markers by overexpressing or knocking down key m6A regulatory proteins, specifically the “writer” methyltransferase METTL3 and the “reader” protein IGF2BP2, in sEVs. We also characterized the mode of action of these proteins via co-immunoprecipitation (co-IP) to elucidate the mechanism by which sEVs mediate m6A modification to regulate HBV replication. We aimed to provide a framework to comprehensively elucidate the mechanisms underlying HBV replication and contribute to the discovery of novel molecular targets for treating HBV infection.
HepG2.2.15 cells (iCell-h093, Shanghai ICell Bioscience Inc., Ltd., Shanghai, China) stably replicating HBV and their parental HepG2 cells (iCell-h092, Shanghai ICell Bioscience Inc., Ltd., Shanghai, China) were cultured in a medium consisting of 8% exosome-free fetal bovine serum (FBS) (Cat #EXO-FBS-50A-1, SBI, Palo Alto, CA, USA) supplemented with 1% penicillin streptomycin (Cat. NO. 15140122, Lot. 238344, Grand Island, NY, USA). To ensure stable HBV replication, HepG2.2.15 medium was supplemented with G418 solution at a concentration of 400 µg/mL (R20001, Lot.NO71R19494A, Shanghai Yuanye Bio-Technology Co., Ltd., Shanghai, China). All cell lines were validated via short tandem repeat (STR) profiling, were tested, and were found to be negative for mycoplasma (Supplementary materials).
After culturing HepG2 and HepG2.2.15 cells for 48–72 h, to remove impurities
such as cells, the cell supernatants were collected and centrifuged at 3000
Initially, 50 µL of HepBsAg (S38: sc-52412, Santa Cruz
Biotechnology, Santa Cruz, CA, USA) was added to the sEVs following
ultracentrifugation. Then, they were transferred to a shaker and placed at 4
°C for 12 h to facilitate thorough rotation and mixing. Next, 20
µL of Protein A/G PLUS-Agarose (sc-22003, Santa Cruz Biotechnology, Santa
Cruz, CA, USA) was added, and the sample was incubated for 8 h at 4 °C
with continuous mixing. After incubation, the mixture was centrifuged at 3000
Purified sEVs from HepG2.2.15 cell culture supernatant (HepG2.2.15-sEVs) and HepG2 cell culture supernatant (HepG2-sEVs) were deposited onto a carbon-coated copper mesh, with 10 µL of each sample added to the grid. After the excess liquid was aspirated from the edges, the samples were dried for 5 min. Next, 10 µL of 2% phosphotungstic acid (Cat# G1870, Lot No. 2400001, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) was added, and the mixture was stained for 1 min. Excess staining solution was removed, and the samples were dried overnight at room temperature. The morphology of the sEVs was observed and photographed using a transmission electron microscope (Hitachi HT7700, Tokyo, Japan).
A 10 µL sample of ultraisolated purified sEVs was added to 1 mL of
1
A 150 µL transfection system for sEVs was prepared using the
Exo-Fect™ Exosome Transfection Reagent Kit (Cat#EXFT20A-1,
Lot.230616-001, SBI, Palo Alto, CA, USA) as follows: 10 µL of Exo-Fect, 20
µL of nucleic acids (20 pmol siRNA or 5 µg plasmid DNA), 50 µL
of purified HepG2.2.15-sEVs, and 70 µL of sterile 1
The sample suspension was centrifuged at 14,000 rpm for 3 min, and the
precipitates were retained. At least 300 µL of sterile 1
Stably grown HepG2 cells were selected and seeded in six-well plates at a
density of 2
Initially, RNA was extracted using the TRIzol (REF.15596026, Lot.257408, Thermo
Fisher Scientific, Waltham, MA, USA) method. After determining its concentration,
the RNA was reverse-transcribed to template cDNA in two steps using an RNA
reverse transcription kit (REF.1622, Lot.3059833, Thermo Fisher Scientific,
Waltham, MA, USA). In step 1, 1 µg of RNA and 1 µL of random primer
were added, followed by the addition of ddH2O to obtain a mixture of 12
µL. The prepared system was reacted at 65 °C for 5 min. In step 2,
4 µL of 5
After extracting RNA using the TRIzol method and determining its concentration, the RNA samples were serially diluted to 200 ng/µL, 100 ng/µL, and 50 ng/µL. The samples were subsequently denatured in a metal bath at 95 °C for 5 min and then cooled immediately on ice. Next, 2 µL of each RNA sample was added dropwise onto a nylon (NC) membrane, which had been cut to the appropriate size. After the RNA was allowed to diffuse naturally and dry, the membrane was cross-linked under UV light for 2 h. Following the cross-linking step, the sealing solution was closed for 1 h.
The NC membrane was incubated at 4 °C for 12–16 h with the primary m6A antibody (dilution 1:1000, Cat. NO. 517924, Lot. NO. KK0512, Chengdu ZEN-BIOSCIENCE Co., Ltd., Chengdu, China). The HRP-labeled goat anti-rabbit IgG secondary antibody (1:5000, Cat.NO. FNSA-0004, Chengdu ZEN-BIOSCIENCE Co., Ltd., Chengdu, China) was added, and the mixture was shaken gently at room temperature for 1 h. The signal was developed using an ECL detection system after washing the film.
An animal tissue/cellular genomic DNA extraction kit was used to extract cellular HBV DNA (Cat# D1700, Lot No. 2311002, Solarbio Science & Technology Co., Ltd., Beijing China), and its concentration was subsequently determined. To digest the HBV DNA, the sample was incubated with T5 nucleic acid exonuclease (D7082S-1, Shanghai Biyuntian Biotechnology Co., Ltd., Shanghai, China) for 30 min at 37 °C. The samples were terminated by adding EDTA and then heat-inactivated at 95 °C for 5 min.
PCR amplification was first performed using primers for HBV DNA, HBV rcDNA, and HBV cccDNA, and then the copy number of the DNA was calculated based on a standard curve. The sequences of primers used were as follows (Sangon Biotech Shanghai Co., Ltd., Shanghai, China): HBV DNA F: 5′-ACCGACCTTGAGGCATACTT-3′, R: 5′-GCCTACAGCCTCCTAGTACA-3′; HBV rcDNA F: 5′-TTTCACCTCTGCCTAATCATCTCT-3′, R: 5′-CTTTATAAGGGTCGATGCCATGC-3′; and HBV cccDNA F: 5′-GCCTATTGATTGATTGGAAAGTATGT-3′, R: 5′-AGCTGAGGCGGTATCTA-3′.
The cells and sEVs were lysed in RIPA buffer containing 1% PMSF (ST507,
Shanghai Biyuntian Biotechnology Co., Ltd., Shanghai, China)
to extract total protein. The samples were lysed on ice for 30 min and
centrifuged at 12,000
After normalizing the protein concentrations, equal amounts of protein were
separated by SDS-PAGE and transferred to 0.45 µm PVDF membranes. Next, 5%
skim milk powder was added for 1 h, after which the appropriate primary antibody
(METTL3, IGF2BP2 primary antibody dilution of 1:1000; Chengdu ZEN-BIOSCIENCE Co.,
Ltd., Chengdu, China) was added, and the membrane was incubated at 4 °C
for 12–16 h. After washing three times with TBST, the membrane was incubated
with an HRP-conjugated secondary antibody (1:5000, Cat.NO. FNSA-0004, Chengdu
ZEN-BIOSCIENCE Co. Ltd., Chengdu, China) at room temperature for 1 h. After the
film was washed, the protein bands were detected using an ECL detection system.
The ImageJ software (Version 1.54v, Java1.8.0, 64-bit; Media Cybernetics, Silver
Springs, MD, USA) was used to analyze the Western blotting bands in grayscale,
and the results were normalized by dividing the target protein grayscale value by
an internal reference grayscale value. The primary antibodies used were as
follows:
After the cells were transfected with sEVs containing the METTL3 plasmid, they were harvested 48 h post-transfection to prepare protein samples. Following the protocol, 20 µL of protein A+G magnetic beads (Cat. NO. P2179S, Lot NO.091322230131, Shanghai Biyuntian Biotechnology Co., Ltd., Shanghai, China) were mixed with 2 µg of either METTL3 primary antibody or normal rabbit IgG and incubated at room temperature for 4 h. Next, protein samples of equal mass were added, and the mixture was rotated overnight at 4 °C.
After incubation, the beads and supernatant were separated using a magnetic rack
for 1 min. Next, 500 µL of a lysis buffer supplemented with protease
inhibitors was used to wash the beads three times. After washing, 50 µL of
1
All data were presented as the mean
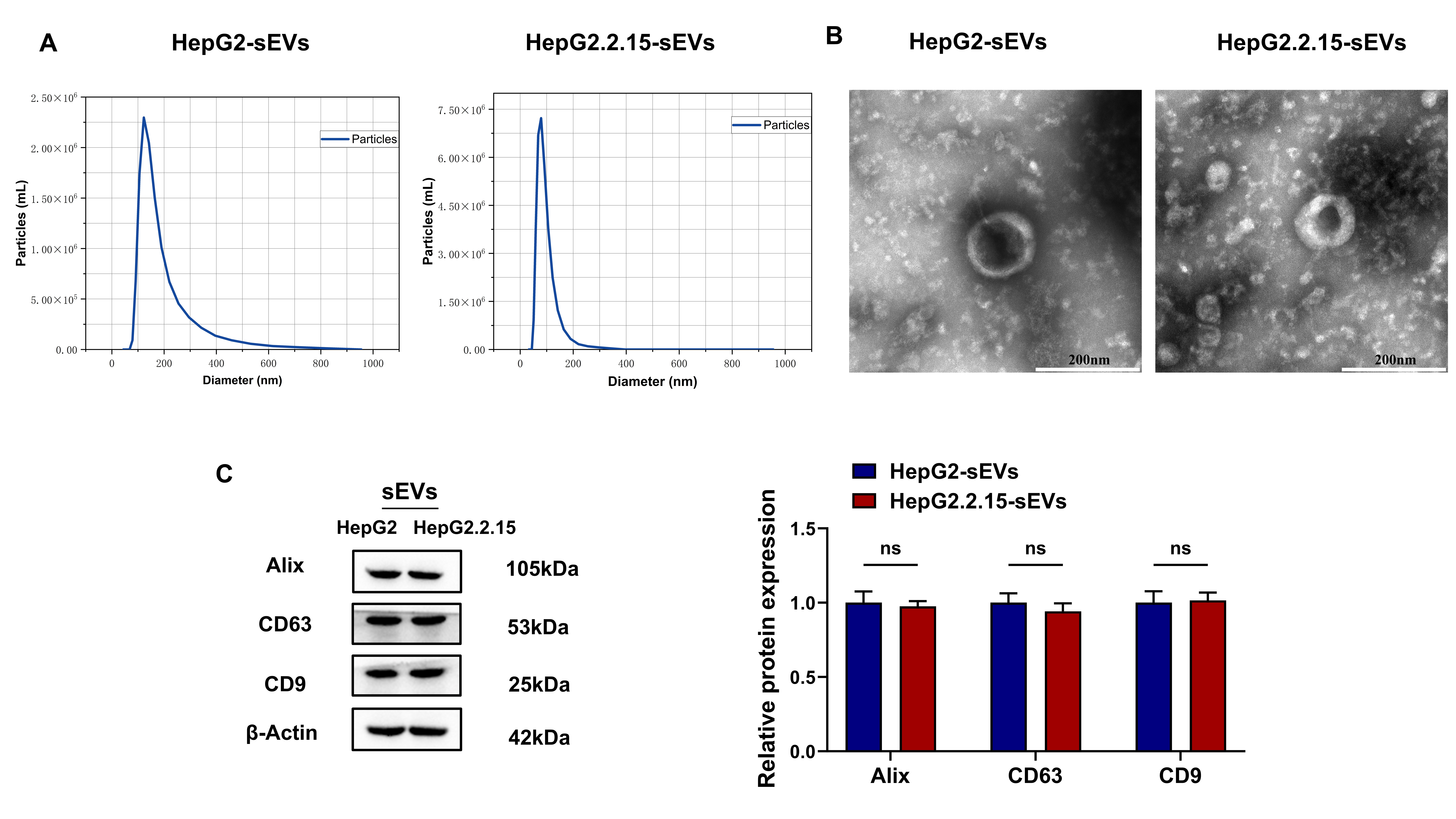
Small extracellular vesicles derived from HepG2 cells (HepG2-sEVs) and
HepG2.2.15 cells (HepG2.2.15-sEVs) were characterized following purification by
ultracentrifugation. DLS analysis revealed that 82.4% of the HepG2-sEV particles
were 30–200 nm in size, and the peak particle size and concentration were 122.4
nm and 2.30
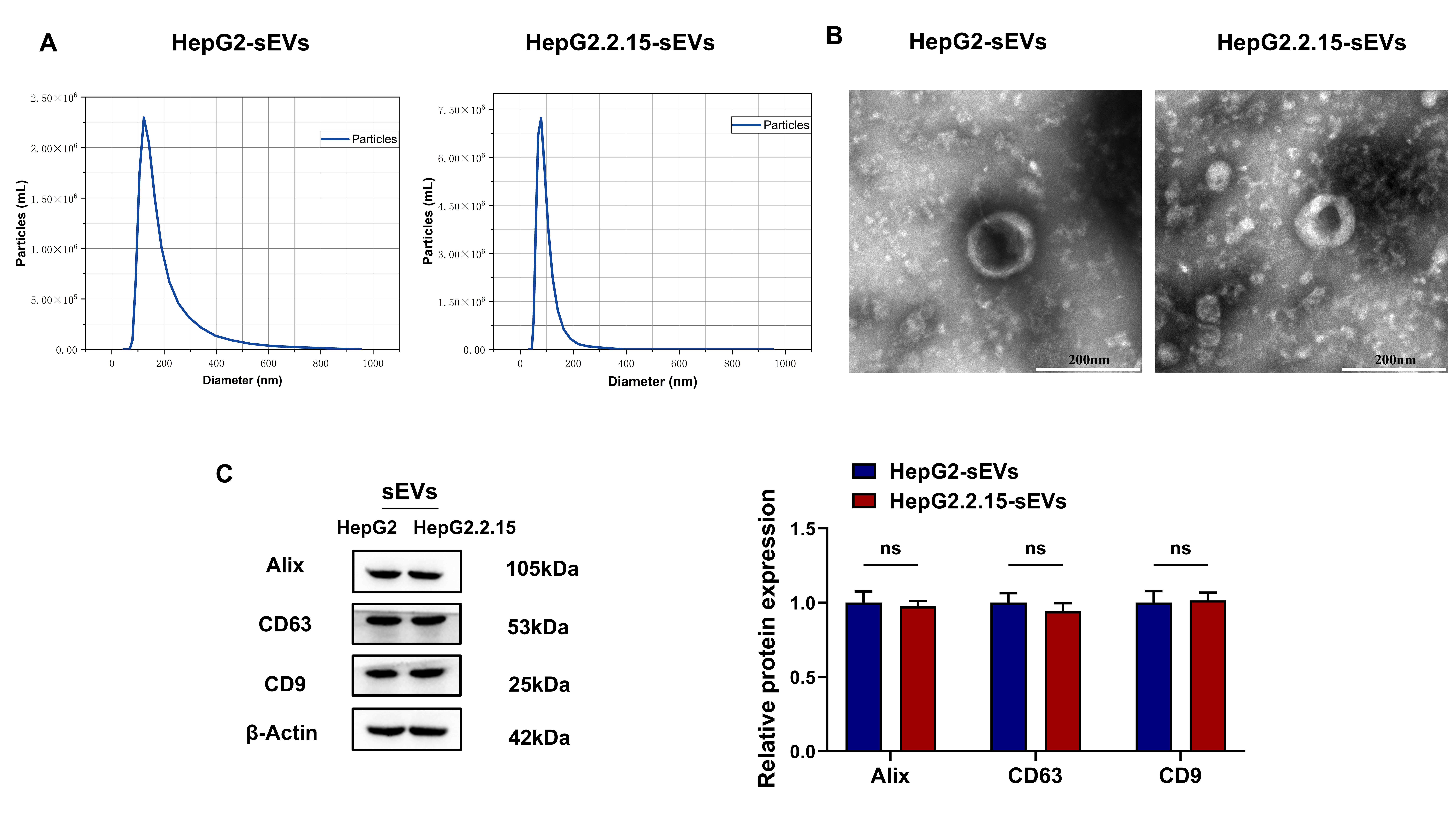 Fig. 1.
Fig. 1.
Identification of sEVs following ultracentrifugation
purification. (A) DLS analysis was conducted to measure the size of sEVs. (B)
TEM imaging was performed to investigate the morphological structure of sEVs. The
scale bar = 200 nm. (C) Western blotting analysis was performed to detect marker
proteins associated with sEVs; ns p
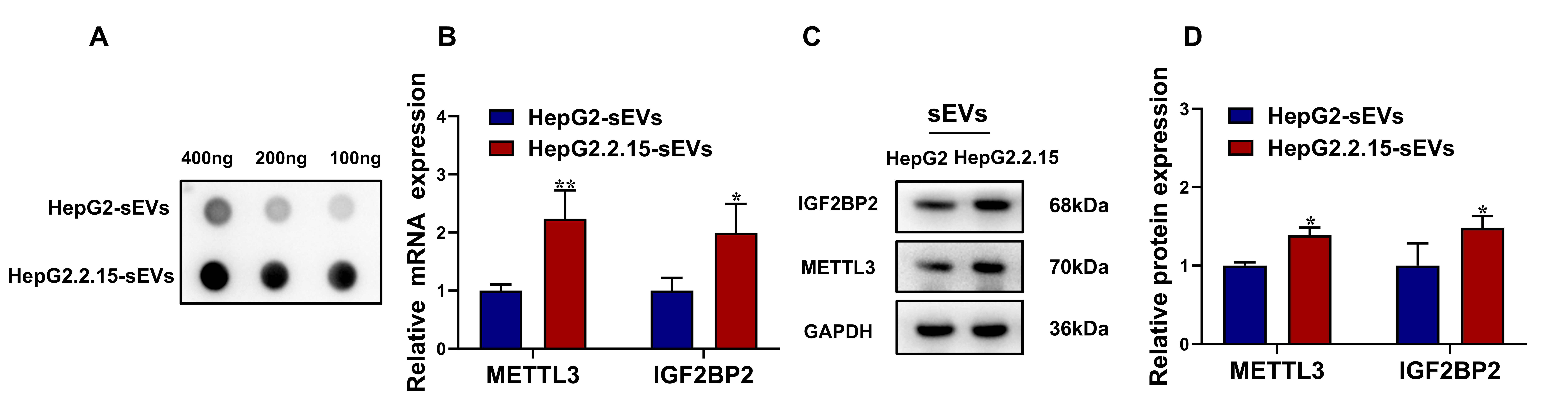
Compared to HepG2-sEVs, HepG2.2.15-sEVs presented significantly greater levels
of m6A methylation (Fig. 2A). The expression levels of METTL3
and IGF2BP2 mRNAs in HepG2.2.15-sEVs were significantly elevated, as
determined by RT-qPCR analysis (Fig. 2B; p
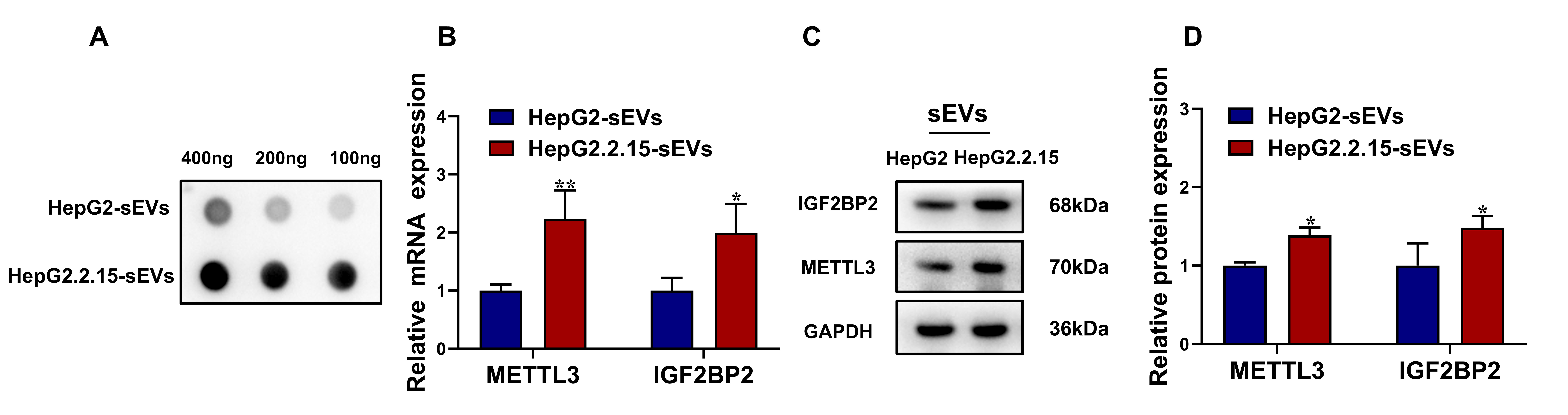 Fig. 2.
Fig. 2.
Enrichment of m6A methylations in sEVs isolated from the
HBV-stably replicating HepG2.2.15 cell line. (A) Dot blotting was performed to
detect the m6A modification levels in the two kinds of sEVs. (B) Quantitative reverse transcription-PCR (RT-qPCR) was
performed to measure the mRNA expression levels of the methyltransferase
METTL3 and the reader protein IGF2BP2 in both types of sEVs.
(C,D) Western blotting was conducted to assess the protein expression levels of
METTL3 and IGF2BP2; *p
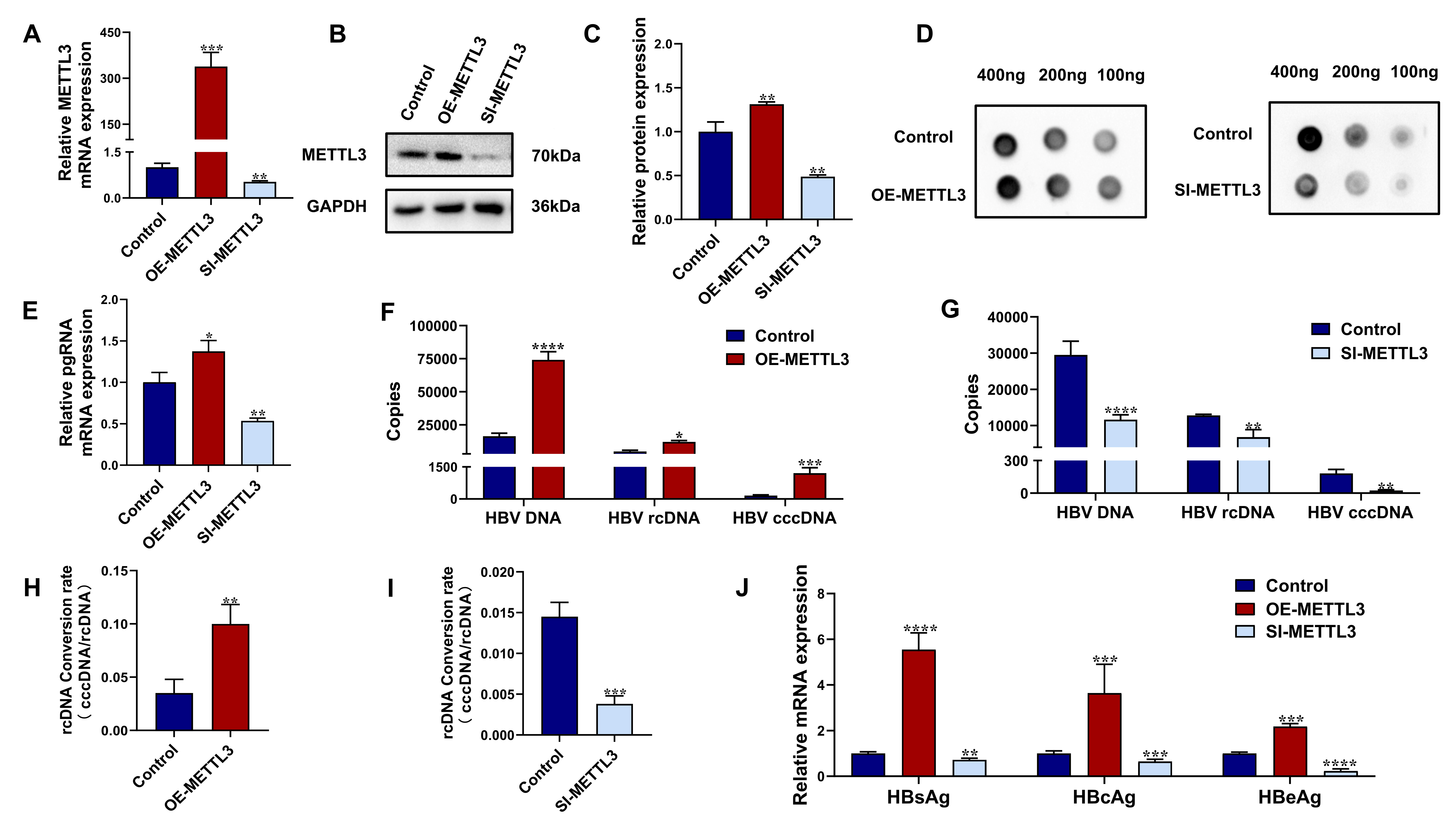
METTL3 was overexpressed or knocked down in HepG2.2.15-sEVs, resulting
in corresponding alterations in METTL3. In the overexpressing METTL3
(OE-METTL3) group, the METTL3 mRNA and protein levels increased.
Conversely, in the SI-METTL3 group,
METTL3 mRNA and protein levels were lower than those in the control
group (Fig. 3A–C; p
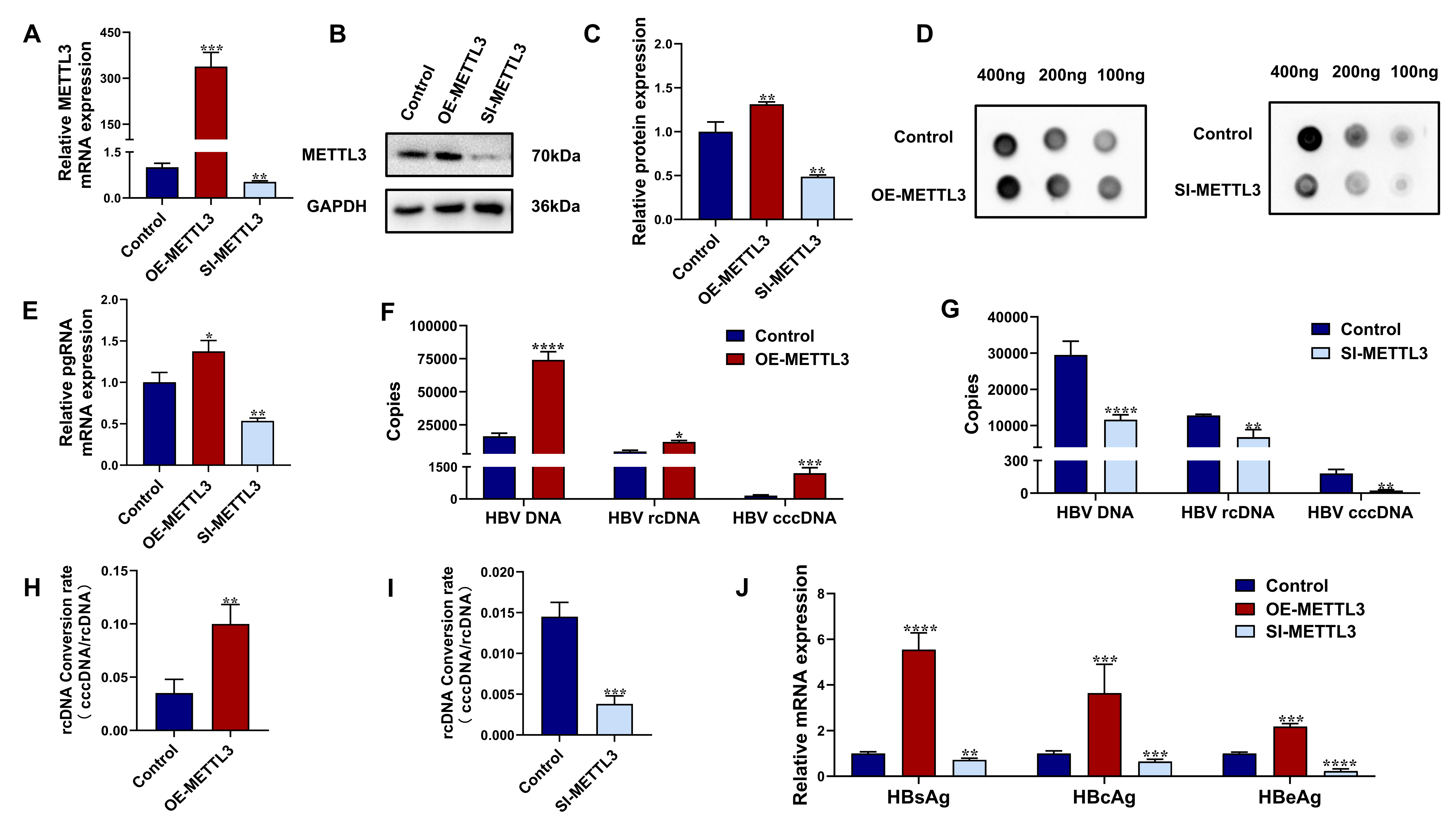 Fig. 3.
Fig. 3.
HepG2.2.15-sEVs enhance HBV replication by upregulating the
methyltransferase METTL3. In the control group,
HepG2.2.15-sEVs without transfection were introduced into parental HepG2 cells.
In the OE-METTL3 group, HepG2.2.15-sEVs overexpressing METTL3 were
transfected into HepG2 cells after transfection with the METTL3 plasmid.
In the SI-METTL3 group, HepG2.2.15-sEVs with METTL3 knockdown were
transfected into HepG2 cells after transfection with METTL3 siRNA. (A)
RT-qPCR was performed to detect the mRNA expression level of METTL3. (B,C) Western blotting analyses were performed to assess the METTL3 protein level.
(D) Dot blot analyses were performed to evaluate the level of m6A modification.
(E–J) qPCR and RT-qPCR analyses were performed to measure the expression rates
of pgRNA, HBV DNA, HBV rcDNA, and HBV cccDNA,the rcDNA conversion rate, and the mRNA expression levels of
HBsAg, HBcAg, and HBeAg;
*p
Compared to that in parental HepG2 control cells treated with untransfected
HepG2.2.15-sEVs, the mRNA level of HBV pgRNA, a marker of HBV
replication, was significantly greater in the OE-METTL3 group (Fig. 3E;
p
In contrast, the SI-METTL3 group presented a decrease in viral replication
indicators: HBV pgRNA expression decreased (Fig. 3E; p
These results suggested that HepG2.2.15-sEVs facilitated an increase in m6A methylation through the upregulation of METTL3, which promoted the conversion of rcDNA to cccDNA, thereby regulating the replication of HBV. Inhibition of METTL3 expression decreased HBV DNA levels in the treatment of HBV-associated diseases, and the detection of METTL3 in sEVs may be used to predict HBV-associated diseases and monitor the prognosis of the disease caused by HBV.
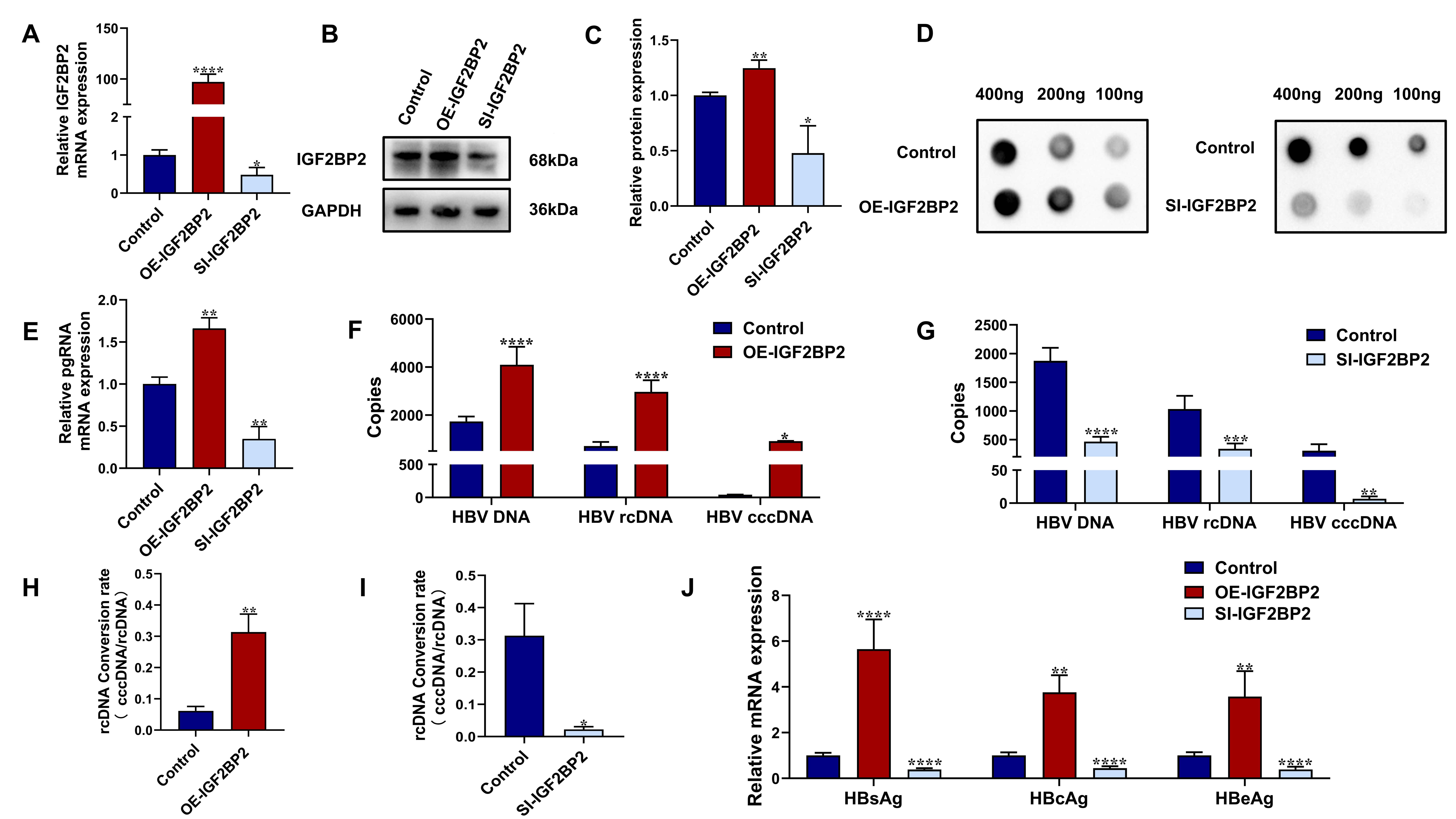
The IGF2BP2 mRNA expression levels increased or decreased (Fig. 4A;
p
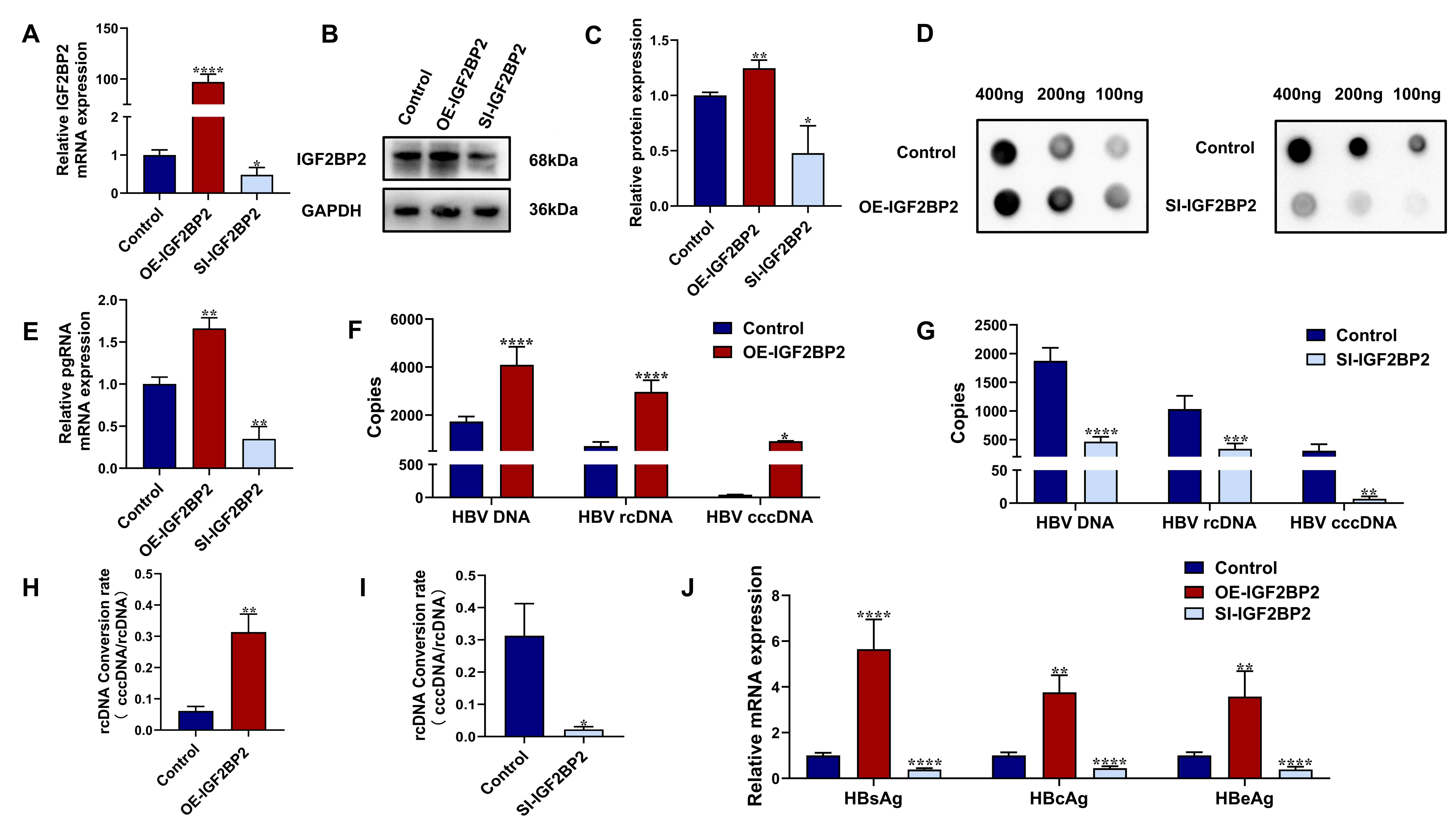 Fig. 4.
Fig. 4.
HepG2.2.15-sEVs enhance HBV replication through IGF2BP2. In the
control group, untransfected HepG2.2.15-sEVs were introduced into HepG2 cells. In
the OE-IGF2BP2 group, HepG2.2.15-sEVs were transfected into HepG2 cells following
transfection with an IGF2BP2 plasmid to induce the overexpression of
IGF2BP2. In the SI-IGF2BP2 group, HepG2.2.15-sEVs were transfected into
HepG2 cells after transfection with IGF2BP2 siRNA to knock down
IGF2BP2. (A) The overexpression or knockdown of IGF2BP2 was
confirmed by RT-qPCR to measure IGF2BP2 mRNA expression. (B,C) The
overexpression or knockdown of IGF2BP2 was confirmed by Western blotting analysis
to assess the level of the IGF2BP2 protein. (D) The level of m6A modification
resulting from the overexpression or knockdown of IGF2BP2 was evaluated
by dot blot analysis. (E–J) The effect of knocking down or
overexpressing IGF2BP2 on HBV replication was assessed through RT-qPCR
and qPCR; the expression levels of pgRNA, HBV DNA, HBV rcDNA,
and HBV cccDNA; the rcDNA conversion rates; and the mRNA
levels of HBsAg, HBcAg, and HBeAg were measured; *p
Compared to the parental HepG2 control cells treated with untransfected
HepG2.2.15-sEVs, the expression level of HBV pgRNA, a marker for
detecting HBV replication, increased or decreased (Fig. 4E; p
The copy numbers of HBV DNA, HBV rcDNA, and HBV cccDNA
increased (Fig. 4F; p
These results suggested that HepG2.2.15-sEVs, via the reader protein IGF2BP2, can increase m6A levels, promote the conversion of rcDNA to cccDNA, and consequently regulate the replication of HBV. A significant reason for the prolonged treatment of HBV disease is the difficulty in eliminating cccDNA. These experimental results suggested that the rcDNA conversion rate may be reduced by inhibiting the expression of IGF2BP2, thus decreasing the generation of cccDNA and halting the progression of the disease.
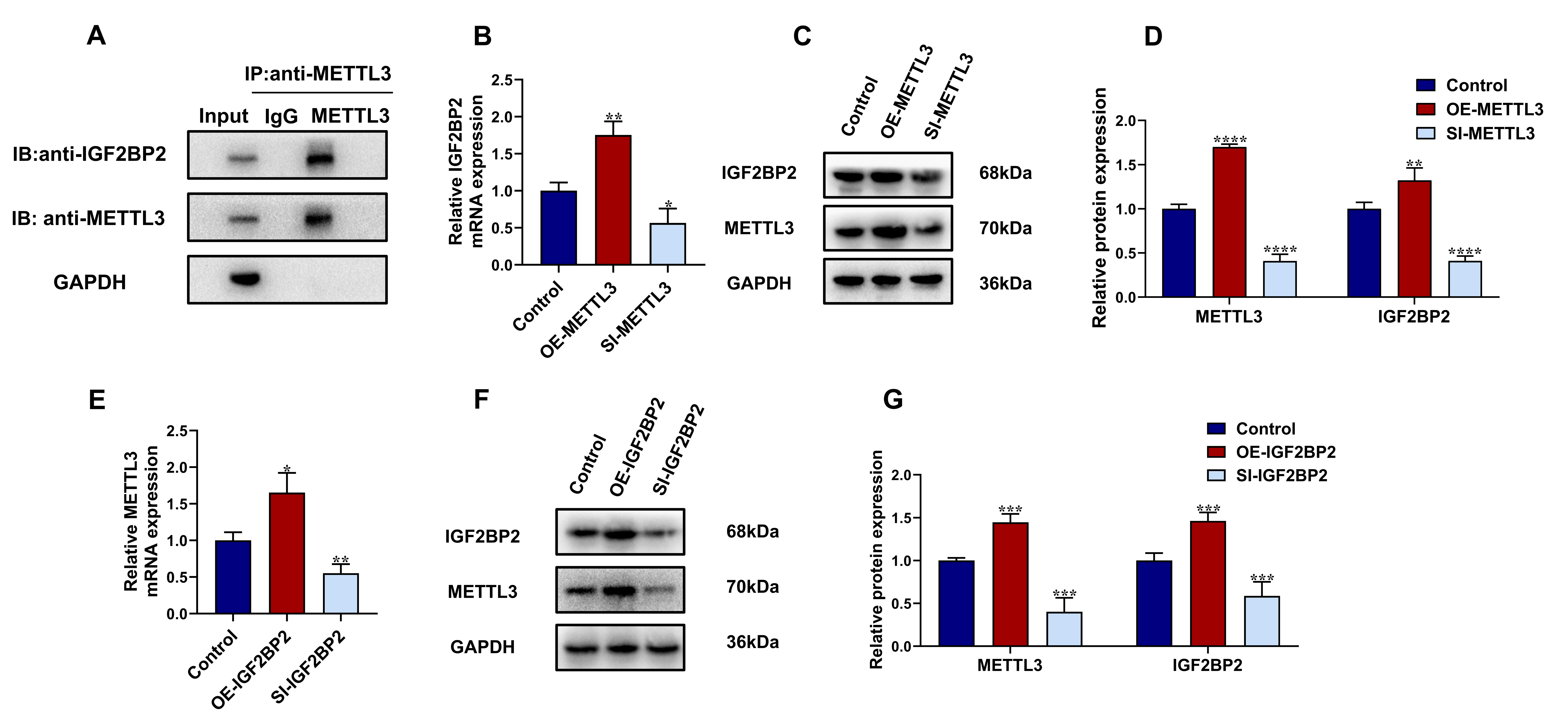
After the cells were transfected with HepG2.2.15-sEVs containing the METTL3 plasmid, IGF2BP2 was detected via immunoprecipitation (IP) using an anti-METTL3 antibody. The immunoprecipitation results indicated a synergistic interaction between METTL3 and IGF2BP2 (Fig. 5A).
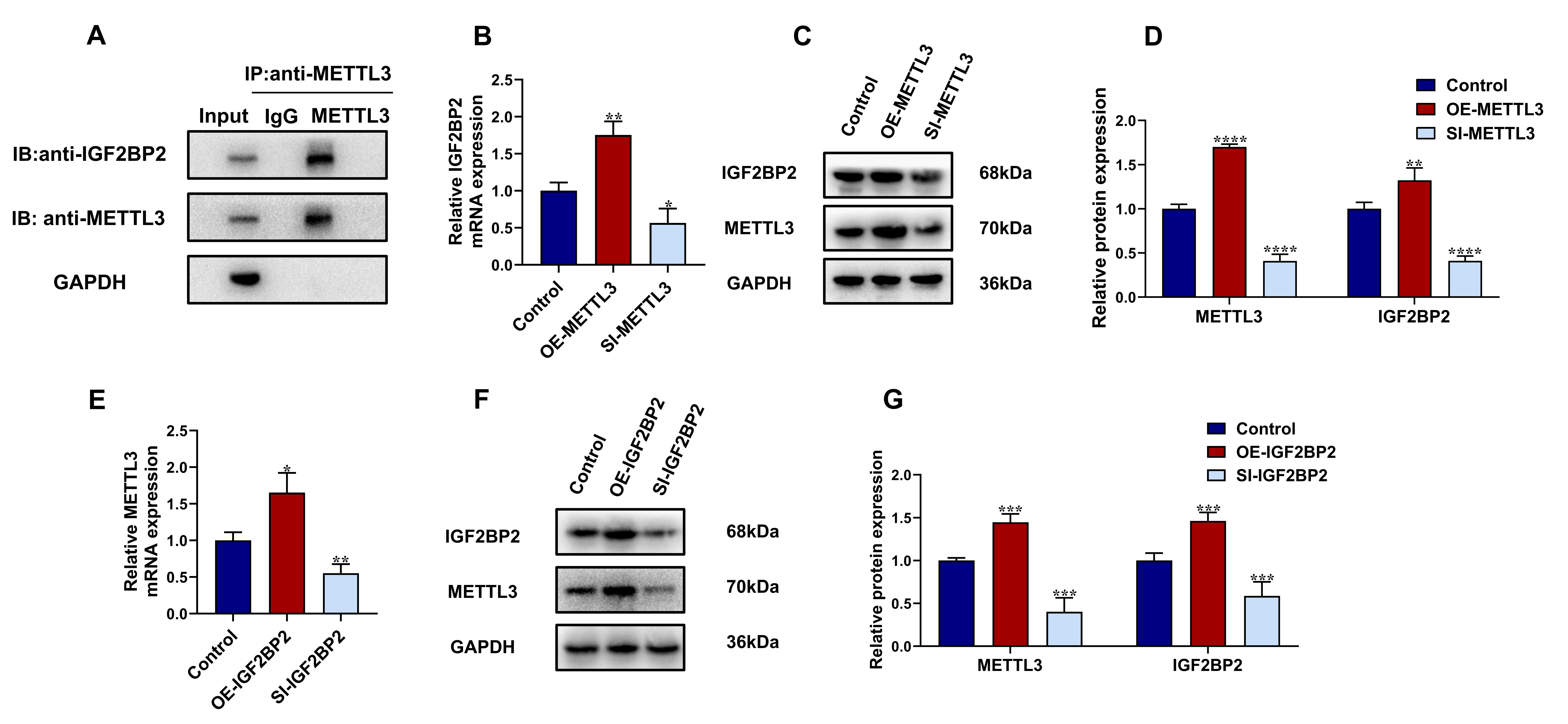 Fig. 5.
Fig. 5.
METTL3 synergizes with IGF2BP2 in
HepG2.2.15-sEVs. The control group consisted of HepG2.2.15-sEVs transfected into
HepG2 cells. The OE-METTL3 group included HepG2.2.15-sEVs overexpressing
METTL3, which were also transfected into HepG2 cells. In the SI-METTL3
group, HepG2.2.15-sEVs with METTL3 knockdown were transfected into HepG2
cells. The OE-IGF2BP2 group included HepG2.2.15-sEVs overexpressing
IGF2BP2, which were transfected into HepG2 cells, whereas the SI-IGF2BP2
group included HepG2.2.15-sEVs with IGF2BP2 knockdown, which were also
transfected into HepG2 cells. (A) Co-IP experiments were conducted to confirm the
interaction between METTL3 and IGF2BP2. (B) Following the overexpression or
knockdown of METTL3, RT-qPCR was performed to assess IGF2BP2
mRNA expression. (C,D) Western blotting analysis was performed to evaluate
METTL3 and IGF2BP2 protein levels after the overexpression or knockdown of
METTL3. (E) RT-qPCR analysis was performed to measure METTL3 mRNA levels
following the overexpression or knockdown of IGF2BP2. (F,G) Western
blotting analysis was conducted to detect the METTL3 and IGF2BP2 proteins
following the overexpression or knockdown of IGF2BP2; *p
After the cells were transfected with HepG2.2.15-sEVs with either overexpression
or knockdown of METTL3, the IGF2BP2 mRNA expression level
increased, as determined by RT-qPCR (Fig. 5B; p
After the cells were transfected with HepG2.2.15-sEVs with either overexpression
or knockdown of IGF2BP2, the METTL3 mRNA expression levels
increased or decreased, respectively (Fig. 5E; p
We previously reported that sEVs carry HBV components that evade immune surveillance, allowing the virus to spread to uninfected cells and promote the replication of HBV [33, 34]. These sEVs possess a stable phospholipid bilayer structure that effectively prevents the degradation of HBV components, and their membrane composition closely resembles that of cellular membranes, facilitating efficient cellular uptake. Among the membrane markers of sEVs, CD63 plays a key role in the packaging, transport, and release of nascent HBV [35]. These findings indicate the importance of sEVs as significant participants and signaling mediators in HBV infection [36, 37].
METTL3-mediated and IGF2BP2-mediated m6A modifications have attracted considerable attention. One study showed that the METTL3-IGF2BP2 complex can promote inflammation through m6A modification, providing important information for the treatment of sepsis [38]. Another study revealed that METTL3 and IGF2BP2 influence the progression of colorectal cancer in a m6A modification-dependent manner [39]. Several studies support the notion that METTL3 and IGF2BP2-mediated m6A modifications play broad roles in various pathogenic mechanisms. Studies have also shown that m6A modifications in HBV RNA may affect its stability and translation, subsequently affecting viral replication [20, 33, 40]. However, the mechanisms by which sEVs mediate epigenetic modifications remain poorly understood. One study indicated that the expression of the methyltransferase METTL3 was significantly higher in the liver tissues of patients with HBV-associated hepatocellular carcinoma compared to normal tissues [41]. This study also suggested that METTL3-mediated m6A modification is crucial for maintaining mRNA stability in an IGF2BP2-dependent manner [42].
Based on these findings, we hypothesized that sEVs mediate m6A epigenetic modifications to promote HBV replication through the METTL3-IGF2BP2 pathway. Our results indicated that METTL3 and IGF2BP2 expression levels were higher in the HBV-stably replicating HepG2.2.15 cell line compared to their expression levels in HepG2 cells [29]. Additionally, m6A modification was significantly enriched in HepG2.2.15-sEVs, suggesting a close association between m6A modification in sEVs and HBV replication.
To elucidate the specific mechanism by which METTL3 and
IGF2BP2 co-mediate m6A modification in sEVs to regulate HBV replication, the
METTL3 gene, a key enzyme, was either overexpressed or knocked down in
HepG2.2.15-sEVs, which were subsequently used to transfect HepG2 cells. The
results indicated a corresponding increase or decrease in m6A methylation, along
with alterations in HBV DNA copy number and HBV pgRNA
expression. These findings suggested that METTL3 facilitates HBV
DNA replication by increasing the m6A modification of HBV mRNAs in sEVs
and increasing pgRNA expression levels. A probable mechanism may be as
follows: METTL3 regulates HBV replication by targeting m6A modification sites
located in the
Transfection of HepG2 cells with HepG2.2.15-sEVs that either overexpress or knock down METTL3 resulted in a considerable increase or decrease in the mRNA levels of HBsAg, HBcAg, and HBeAg. These findings suggested that METTL3 within sEVs influences the transcription of HBV core proteins and cytosolic proteins, thus leading to an increase in mRNA expression levels. This regulation affects HBV viral packaging and promotes viral replication. A similar observation was reported in a related study by Cheng et al. [43]. Furthermore, an examination of the HBV viral genomic rcDNA and cccDNA revealed that the copy numbers of rcDNA and cccDNA changed simultaneously, accompanied by a significant alteration in the rcDNA to cccDNA conversion rate. These findings indicated that METTL3 in sEVs enhances HBV replication by increasing the expression of HBV rcDNA and cccDNA, as well as by facilitating the conversion of rcDNA to cccDNA, which in turn supports the replication of HBV [1, 2, 44].
We found that sEVs-mediated METTL3 enhances m6A methylation of HBV mRNA and pgRNA, resulting in an increase in the levels of HBV DNA and promoting the conversion of rcDNA to cccDNA. This process serves as a template that enhances the transcription of core viral proteins (HBsAg, HBcAg, and HBeAg), ultimately regulating the replication and transcription of HBV.
Further experiments involving the overexpression or knockdown of IGF2BP2 in sEVs revealed a significant increase or decrease in the copy number of HBV DNA, indicating that IGF2BP2 also plays a role in regulating HBV replication within sEVs. This finding aligned with observations at the cellular level that a targeted reduction in IGF2BP2 inhibits the proliferation of HepG2.215 cells and viral replication [45]. To elucidate the mechanism underlying the role of IGF2BP2 in sEVs, the copy numbers of rcDNA and cccDNA were also examined. The results indicated that overexpressing or knocking down IGF2BP2 in sEVs increased or decreased rcDNA and cccDNA copy numbers in transfected HepG2 cells, along with a corresponding increase or decrease in the rcDNA to cccDNA conversion rate. These findings suggested that IGF2BP2 enhances the expression of rcDNA and cccDNA and promotes the conversion of rcDNA to cccDNA, thereby facilitating the replication of HBV.
The overexpression or knockdown of IGF2BP2 in sEVs resulted in an increase or decrease in the mRNA expression levels of pgRNA, HBsAg, HBcAg, and HBeAg, respectively, along with corresponding alterations in m6A methylation levels in transfected HepG2 cells. These findings indicated that IGF2BP2 in sEVs mediates m6A modifications to positively regulate the transcription of HBV RNA. IGF2BPs can recognize GGm6AC motifs, thus promoting mRNA translation and enhancing mRNA stability [46, 47, 48]. Based on these findings, we speculated that IGF2BP2 in sEVs binds to HBV mRNA, enhancing its stability and promoting translation. This interaction increases mRNA expression of HBV core proteins and cytosolic proteins and also elevates viral DNA levels. This process also facilitates the conversion of rcDNA to cccDNA, thereby regulating the replication and transcription of HBV. Although functionally similar to METTL3, the underlying mechanisms probably differ.
Therefore, the mechanism of action of METTL3 and IGF2BP2 is critical. After METTL3 and IGF2BP2 in sEVs were overexpressed or knocked down, we found a corresponding increase or decrease in the expression levels of IGF2BP2 and METTL3 in the transfected cells. Co-IP experiments showed a synergistic interaction between METTL3 and IGF2BP2. These findings suggested that sEVs regulate HBV replication through the collaborative interaction of METTL3 and IGF2BP2, which mediates m6A methylation. Some studies have also shown that the METTL3-IGF2BP2 axis can interact to regulate m6A methylation [21, 49, 50]. The methyltransferase METTL3 and the reading protein IGF2BP2 work synergistically to form a complex; the process involved is intricate and involves multiple molecules. Among these molecules, the most notable players include IGF2BP1 and IGF2BP3 from the IGF2BP family, which frequently bind to IGF2BP2 [21]. Therefore, we hypothesized that they may also play a role in the synergistic action of METTL3 and IGF2BP2. However, this hypothesis needs to be experimentally verified.
In this study, we investigated the mechanisms regulating HBV replication, beginning at the molecular level of sEVs, while integrating the contemporary research interest in m6A methylation (Fig. 6). This approach offers a relatively novel perspective; however, other genetic components and regulatory pathways may also be involved. For example, IGF2BP2 can directly bind to the structure-specific nuclease Flap Structure-Specific Endonuclease 1 (FEN-1) mRNA, thereby influencing the stability of FEN-1, which presents an additional avenue for future research. However, our study had some limitations, including the absence of certain in vivo experimental data. Therefore, further investigations are needed, and we aim to assess these avenues in our future study.
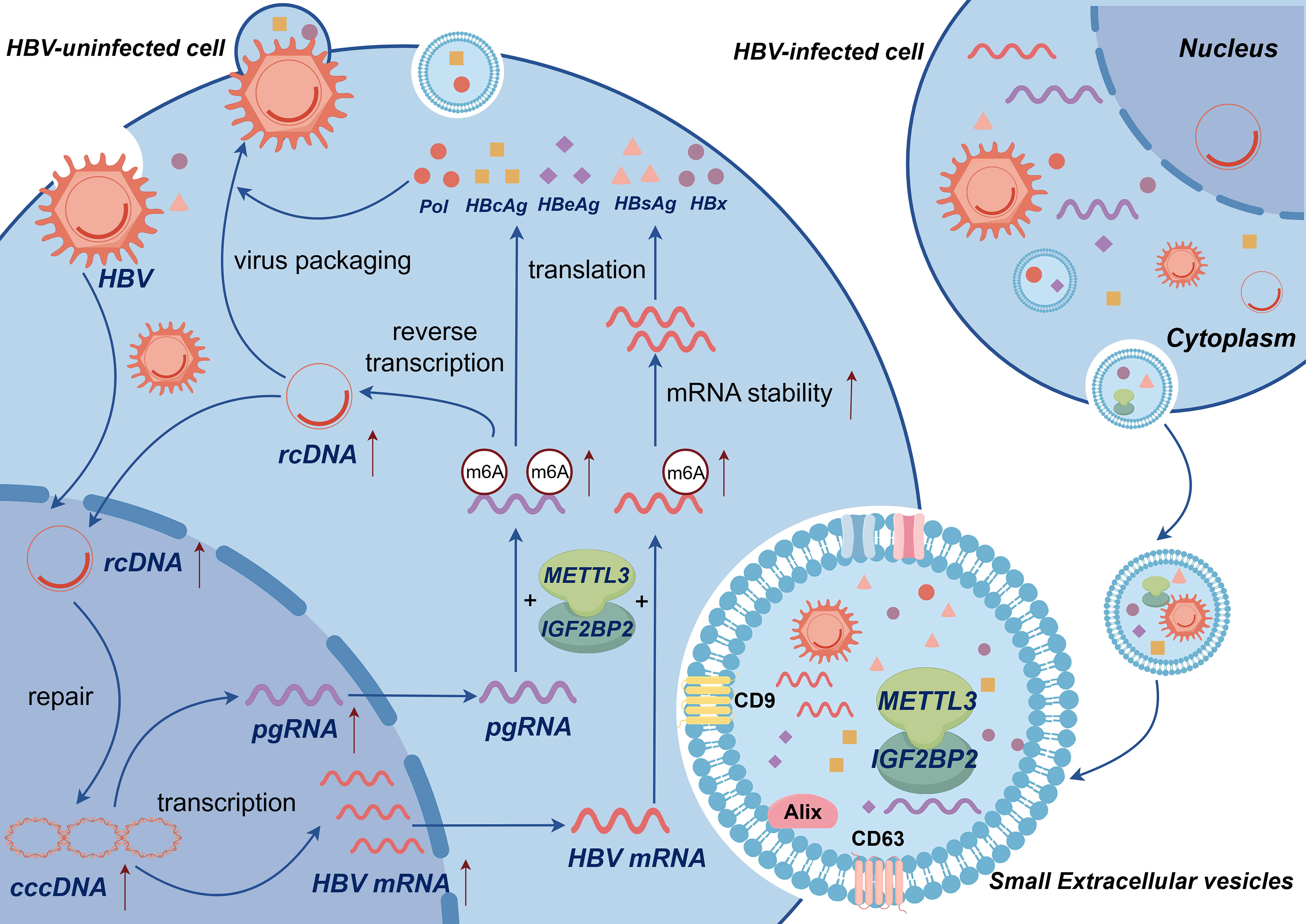
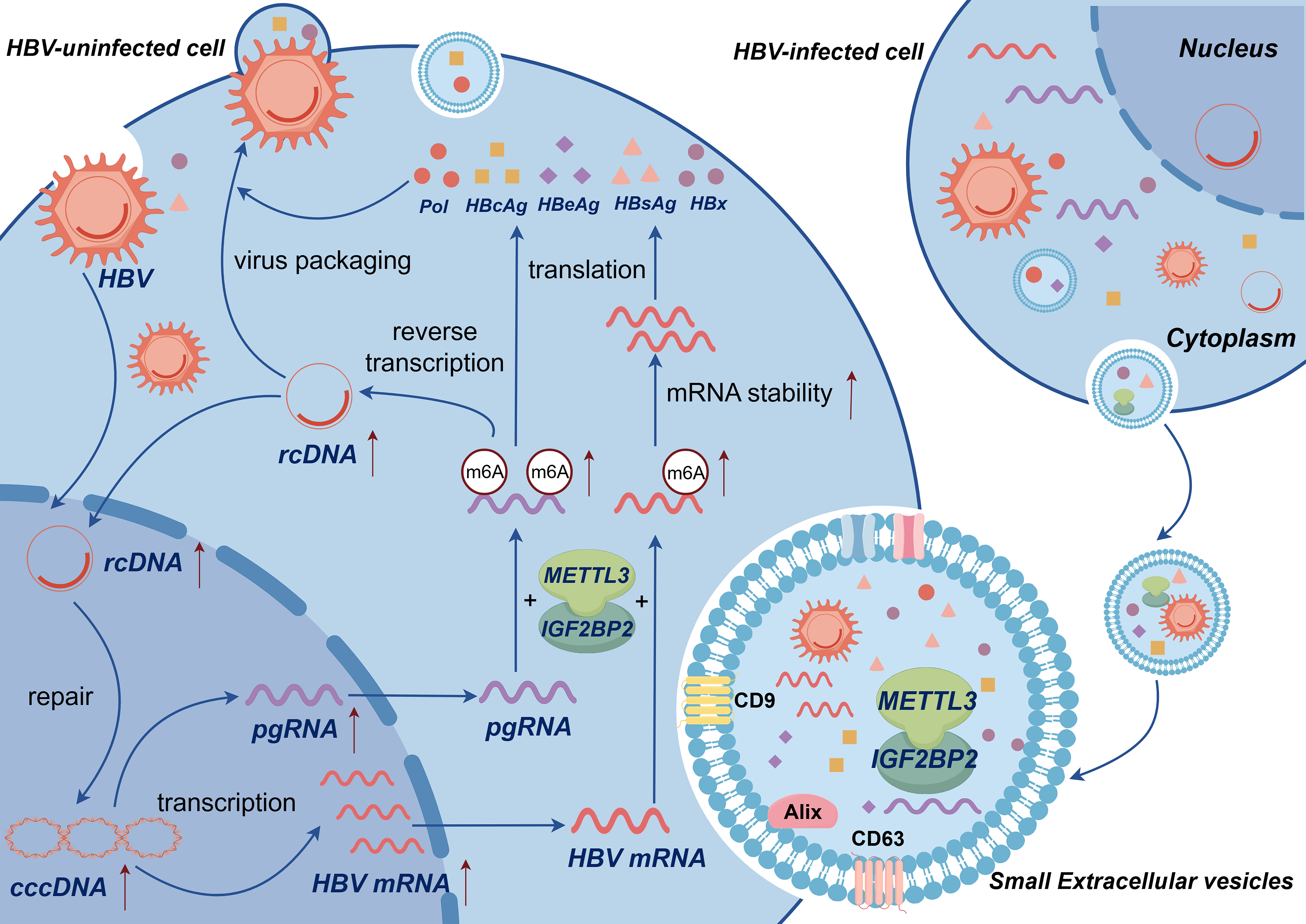 Fig. 6.
Fig. 6.
Mechanistic model in which METTL3 and IGF2BP2 synergistically mediate m6A modification to regulate HBV replication. Small extracellular vesicles (sEVs) enhance m6A epigenetic modification through the coordinated action of METTL3 and IGF2BP2. This process facilitates the conversion of HBV rcDNA to cccDNA, increases the level and stability of HBV core gene mRNA transcripts, including pgRNA, HBsAg, HBcAg, and HBeAg, and ultimately regulates the replication and transcription of HBV (Source: Figdraw 2.0, a platform from China affiliated with the House of Researchers).
To summarize, sEVs enhance m6A epigenetic modification through the synergistic interaction of METTL3 and IGF2BP2, promoting the conversion of HBV rcDNA to cccDNA and increasing the mRNA transcript levels of HBV core genes, including pgRNA, HBsAg, HBcAg, and HBeAg. This process regulates HBV replication and transcription. In this study, we investigated the molecular mechanism by which METTL3 and IGF2BP2 collaborate to mediate m6A methylation in sEVs, thereby regulating the replication of HBV. However, the effect of sEVs and their contents on HBV regulation necessitates further validation through additional studies. These findings may serve as a basis for developing new therapeutic targets and pathways for treating HBV infection.
The datasets used during the current study are available from the corresponding author on reasonable request.
JZ and WP designed the research study, JZ performed the research and analyzed the data, LY and XW gave guidance on experimental techniques. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Sichuan North Medical College for providing the platform to complete the experiments and Nanchong Municipal Science and Technology Strategic Cooperation Program with Colleges and Universities for the financial support.
The work was supported by the Special Project of Strategic Cooperation in Science and Technology of Nanchong City and Colleges and Universities (22SXQT0318).
The authors declare no conflict of interest.
Supplementary materials associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL36291.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.