
The respiratory rhythm is generated by the interaction of oscillators disparately distributed throughout the pons, medulla, and spinal cord. According to the classic model, the interaction amongst preBötzinger complex (preBötzC) spontaneously bursting preinspiratory units and Bötzinger complex (BötzC) expiratory cells generates the principal respiratory rhythm, thence relayed caudally to the pattern generating elements and premotoneurons of the rostral and caudal divisions of the ventral respiratory group and bulbospinal units of the dorsal respiratory group. Rhythm and pattern generating elements in the ventrolateral medulla receive powerful phasic and tonic modulatory inputs from diencephalic structures, midbrain, Kölliker-Fuse, and parabrachial nuclei, retrotrapezoid nucleus, parafacial respiratory group, ventrolateral metencephalon, nucleus tractus solitarius, and brainstem reticular formation, collectively shaping the normal eupneic discharge. Empirical and computational studies have generated models of respiratory rhythmogenesis and pattern formation variously predicated upon pacemaker, network, or hybrid pacemaker network mechanisms to explain oscillatory behavior and regularity. Network mechanisms critically require the integrity and functionality of inhibitory synaptic neurotransmission. The operation and contribution of inhibitory elements in respiratory rhythm generation and pattern formation are well demonstrated empirically and incorporated in computational network and hybrid models of breathing. Fast inhibitory synaptic neurotransmission utilizes GABAAergic and glycinergic mediated activation of receptor linked chloride conductances, generating an inwardly directed flux of chloride ions mediating membrane voltage hyperpolarization and is required to generate eupneic respiratory patterns in vivo and situ. Persistence of rhythmicity in the presence of synaptic antagonism of GABAA and glycine receptor mediated fast inhibitory neurotransmission indicates pacemaker generating mechanisms sufficiently capable of independently generating this behavior in vitro and transected intact preparations maintaining the preBötzC as the most rostrally preserved structure. The role of GABAB receptor mediated neuromodulation in respiratory rhythm generation and pattern formation is comparatively significantly less investigated. GABABergic activation of postsynaptic and presynaptic membrane receptors variably upregulates potassium conductances and downregulates calcium conductances. Respiratory rhythm and pattern are powerfully modulated in vivo, in situ, and in vitro by superfusion or localized microinjections of GABABergic agonists and antagonists, though are typically not abolished by these experimental interventions. Directionality and magnitude of these effects exhibit maturational changes. The relative depolarization of chloride reversal potentials during the early neonatal period, with gradual shifts towards normal hyperpolarizing values during development, suggests GABABergic signaling may mediate the inhibitory neurotransmission necessary to generate triphasic eupnea. We review and discuss the role of spontaneously bursting oscillators and network mechanisms predicating upon fast inhibitory synaptic neurotransmission in contributing to respiratory rhythmogenesis and pattern formation.
Breathing sustains normal gas exchange and life. Triphasic eupnea consists of neural inspiratory (I), postinspiratory (post-I), and late expiratory (late-E) phases, as characterized by the pioneering works of Richter and colleagues (Richter et al., 1986), a definition having withstood the test of time for the past several decades (Anderson and Ramirez, 2017; Garcia et al., 2019; Morgado-Valle and Beltran-Parrazal, 2017; Ramirez and Baertsch, 2019). Respiratory rhythm generation thus represents bursting regularity, and pattern formation represents the coordinated spatiotemporal organization of inspiratory, post-inspiratory, and expiratory neural output effector mechanisms. The biophysical properties of the rhythm generator determine burst onset, offset, and frequency. The elements of the pattern generator shape burst content. The neural respiratory output is conveyed to and drives the muscles of inspiration and expiration. The challenges of central pattern generation include the genesis of consistently reproducible rhythmic activity and an organized pattern coordinating inspiratory and expiratory motor outputs, responsiveness to lung stretch and hypercapnic and hypoxic chemosensory demands, integration of neural respiratory output with sympathetic and parasympathetic outflows, integration with the behavioral demands of the animal, and coordination with other rhythmic activities (Molkov et al., 2017).
The core rhythm generating neuronal circuitry of the medullary preBötzinger (preBötzC) (Morgado-Valle and Beltran-Parrazal, 2017) and Bötzinger (BötzC) (Ezure and Manabe, 1988) complexes and pattern generating neuronal elements of the ventral (VRG) (Ellenberger and Feldman, 1990; Ghali, 2017c; Hayakawa et al., 2004) and dorsal respiratory groups (DRG) (Ghali, 2017c; Lipski et al., 1990; Richardson and Mitchell, 1982; Voss et al., 1990) interact to generate alternating inspiratory expiratory rhythmic activity (Molkov et al., 2017) (Figs. 1 and 2). The preBötzC contains preinspiratory (pre-I), preinspiratory inspiratory (pre-I/I) phase spanning units, and decrementing early inspiratory (dec early-I) units variably exhibiting intrinsic bursting properties (Morgado-Valle and Beltran-Parrazal, 2017) and represents the principal kernel of inspiratory rhythm generation (Marchenko et al., 2016; Smith et al., 1991). This zone was identified as the rostralmost structure requiring preservation in order to maintain the generation of a respiratory rhythm exhibiting monophasic inspiratory discharge, elegantly demonstrated by the seminal work of Smith and colleagues (Smith et al., 1991) in the in vitro preparation of the neonatal rat. The BötzC contains decrementing postinspiratory (dec post-I) (Ezure and Manabe, 1988) and augmenting late expiratory (aug late-E) (Jiang and Lipski, 1990) neurons providing powerful inhibition to preBötzC pre-I units and the rostral ventral respiratory group (rVRG) augmenting inspiratory (aug-I) cell population (Yang and Feldman, 2018) and driving the expiratory premotoneurons of the caudal ventral respiratory group (cVRG) (Ezure et al., 2003; Long and Duffin, 1986; Molkov et al., 2017; See et al., 1987; Shen et al., 2003; Tonkovic-Capin et al., 2003).
 Figure 1.
Figure 1.Conceptual model of the brainstem drive to the phrenic motor network. Color traces under phrenic neurograms indicate phase of activity (i.e., inspiratory, expiratory [post-I and E2], or tonically discharging units) of indicated excitatory (+) and inhibitory (-) synapses Medullary drive to phrenic motor nucleus. The retrotrapezoid nucleus is a central chemoreceptor region with exquisitely chemosensitive glutamatergic units. These cells are robustly stimulated by acid, exhibiting a steep slope of neuronal discharge frequency versus concentration of hydrogen ions. Hydrogen ions are generated principally from the reaction of arterial CO2 with H2O generating the weak acid H2CO3. The weak acid H2CO3 is in equilibrium with its dissociation products H+ cation and HCO3- cation. The HCO3- anion dissociates into H+ and CO32-, with an equilibrium preferentially favoring the formation of HCO3-. The equilibria and dynamics are governed by the Henderson-Hasselbach equation, Ph = pKa + log $\frac{[H^+][A^-]}{[HA]}$. RTN glutamatergic units provide prominent tonic excitatory drive and support to the Bötzinger and pre-Bötzinger complexes, ventral respiratory column nuclei, dorsal respiratory group, and pontine elements constituting the brainstem neural respiratory network. The Bötzinger and pre-Bötzinger complexes interact to generate a core two-phase neural respiratory rhythm and control premotoneurons in the rostral and caudal divisions of the ventral respiratory group. BötzC dec post-I and aug late-E units provide inhibition to preBötzC pre-I, pre-I/I, and dec early-I units and propriobulbar excitatory drive to cVRG expiratory premotoneurons. PreBötzC units reciprocally inhibit BötzC expiratory cells. PreBötzC excitatory pre-I and pre-I/I units and drive rVRG aug-I units and preBötzC inhibitory dec early-I units shape inspiratory ramp by inhibiting rVRG aug-I premotoneurons during early inspiration. Rostral ventral respiratory group premotoneurons drive the phasic activity of phrenic motoneurons through projections conveyed through ipsilateral (pathways which either do not decussate or decussate twice at the medullary level then upper C1-C2 cervical spinal cord or phrenic motoneuronal level) and contralateral (pathways which decussate once at either the brainstem, upper cervical spinal cord, or phrenic motor nucleus levels) ventromedial and lateral funiculi of the spinal cord relaying to phrenic motoneurons monosynaptically or through an interposed pre-phrenic interneuron located in the upper cervical spinal cord or phrenic motor nucleus. Phrenic motoneuronal dendrites projecting into the contralateral hemicord may receivie descending inputs from rostral ventral respiratory group conveyed through the ventromedial and lateral funiculi of the spinal cord. Phrenic motoneurons with dendrites decussating across the midline represent a significant fraction of these units during early neonatal age and evidence rapid age-dependent decreases. Medullophrenic units (primarily from the BötziC) contribute to phasic inhibition of PhMNs. Local pre-phrenic interneurons may also convey phasic inhibition and contribute to tonic inhibition of PhMNs. RTN, retrotrapezoid nucleus; BötzC, Bötzinger complex; pre-BötzC, pre-Bötzinger complex; rVRG, rostral ventral respiratory group; cVRG, caudal ventral respiratory group; C1-C2 pre-PhINs, C1-C2 pre-phrenic interneurons; PhNucl, phrenic nucleus; PhL, left phrenic nerve; PhR, right phrenic nerve. Modified with permission from Fig. 10 Ghali and Marchenko (2016a).
 Figure 2.
Figure 2.Pontomedullary neural respiratory oscillators. The pontomedullary ventral respiratory column (VRC) nuclei provide the neuroanatomic substrate generating the respiratory rhythm. The ventrolateral column of pontomedullary nuclei generating the respiratory rhythm and pattern, progressing from rostral to caudal are comprised by and include the retrotrapezoid nucleus/parafacial respiratory group (RTN/pFRG), in the immediate vicinity of and bordering the facial nucleus (VII); the postinspiratory complex (PiCo), located caudal to the facial nucleus and dorsomedial with respect to the nucleus ambiguus (NA); the Bötzinger (BötC) and preBötzinger complexes (preBötC), located ventromedial to the nucleus ambiguus; and the rostral (rVRG) and caudal (cVRG) divisions of the ventral respiratory groups, located deep and dorsal to the lateral reticular nucleus (LRt). The borders of the compartments containing the ventral respiratory column nuclei are indistinct. Electrophysiological properties and differential spatiotemporal dynamics of discharge have characteristically distinguished these regions, though recent studies have identified specific genetic and molecular markers identifying these zones. Modified with permission from Fig. 1 of Anderson et al. (2016).
In brief, the provision of tonic and phasic excitatory and inhibitory inputs deriving from pontine nuclei, including the Kölliker-Fuse and medial parabrachial nuclei (Dutschmann and Herbert, 2006; Mörschel and Dutschmann, 2009), as well as the retrotrapezoid nucleus (RTN) (Guyenet and Mulkey, 2010; Guyenet et al., 2019) and the parafacial respiratory group (pFRG) oscillator (Onimaru and Dutschmann, 2012; Onimaru et al., 1987, 1988) shapes the rhythmic activity into smooth triphasic eupnea, comprised of central neural I, post-I, and late-E activities (Ghali, 2015; Ghali and Marchenko, 2016b; Richter et al., 1986). Rostral VRG aug-I premotoneurons (Stornetta et al., 2003) drive the rhythmic discharge of inspiratory networks in brainstem and cervicothoracic motor nuclei (Ellenberger and Feldman, 1990; Ghali and Marchenko, 2013, 2015, 2016a,b; Ghali, 2017c,d; Ghali, 2018; Marchenko et al., 2012; Zaki Ghali et al., 2019;) and cVRG expiratory premotoneurons (Tonkovic-Capin et al., 2003) drive the discharge of expiratory networks in brainstem and abdominal motor nuclei through direct monosynaptic (Ghali, 2017c) and indirect polysynaptic interneuronal relays (Molkov et al., 2017; Zaki Ghali et al., 2019). Respiratory rhythm generation models are based either on the activity of pacemaker cells (Morgado-Valle and Beltran-Parrazal, 2017), network activity (Molkov et al., 2017; Rybak et al., 2014; Smith et al., 2000. 2007, 2009), or hybrid combinations thereof. Network mechanisms require the integrity and functionality of fast inhibitory synaptic neurotransmission, representing a critically debated subject amongst authors seeking to illumine mechanisms generating the breathing rhythm. The network generating the breathing rhythm and pattern is significantly more complex than the description presented herein and exceptionally reviewed by Anderson and Ramirez (2017). The brainstem network generating breathing exhibits significant neuroanatomic and functional overlap with the networks generating sympathetic and parasympathetic oscillations (Ghali, 2017a,b,c,d).
Respiratory central pattern generators (CPGs) operating in different modes and network reconfigurations, thus allow adaptation to respiratory stressors under different conditions. In this regard, the respiratory CPG is subject to powerful modulation by descending inputs from the cerebrum, midbrain, and cerebellum (Horn and Waldrop, 1997) and peripheral inputs from hypercapnia, hypoxia, and lung stretch (Ghali and Marchenko, 2016b; Guyenet et al., 2019). Hypercapnia stimulates central (e.g., RTN, nucleus tractus solitarius [NTS], raphe nuclei) and peripheral chemoreceptors (e.g., glomus cells of the carotid bodies and aortic arch, retroperitoneum) in order to appropriately augment the activity of neurons within the ventral respiratory column nuclei. Hypoxia generates a biphasic ventilatory response, with initial augmentation of breathing, mediated preferentially through the stimulation of peripheral chemoreceptors (e.g., glomus cells of carotid bodies and aortic arch), though central hypoxic chemosensitivity is also well described (e.g., preBötzC, NTS, fastigial nucleus), followed by hypoxic ventilatory depression, mediated by rubral modulatory active inhibition principally conveyed monosynaptically to medullary respiratory-related units (Ghali, 2019b; Schmid and Bohmer, 1988; Waites et al., 1996). Pump cells of the NTS project a GABAergic modulatory influence upon chemosensitive RTN units (Marchenko and Rogers, 2007a; Takakura et al., 2007).
The works of Flourens in 1858 would identify the medulla as the nœud vitale of breathing principally generative of the respiratory rhythm (Flourens, 1858). Development of our convenient designation of the medullary respiratory circuitry into discrete ventral respiratory column nuclei and dorsal respiratory groups is predicated upon identification of unique patterns of neuronal discharge by authors performing contemporaneous recordings of individual units in medulla and respiratory-related neurograms, derived from the works of myriad investigators, including C. Von Euler, R.W. Richter, and M.I. Cohen. Mitchell and Berger (1975) were the first authors to use the term ventral respiratory group in a state of the art review discussing the mechanisms generating breathing. These experiments essentially represented neurophysiological fishing expeditions in the medulla of anesthetized and unanesthetized decerebrate cats, rabbits, and rats. Authors spatiotemporally designated patterns of the unitary discharge as they related to recordings of inspiratory and expiratory related neurograms and locoregionally characterized patterned clusters of respiratory-related unitary discharge and responses to various peripheral perturbations (e.g., chemoreceptor stimulation, pulmonary stretch loading and unloading). Antidromic stimulation and collision tests identified the neurocellular architecture of respiratory-related propriobulbar, bulbospinal, and motor neurons. Cross-correlation and fast Fourier transformation and time-frequency spectral and coherence analyses of respiratory-related neurograms identified correlated activities and signatures of the respiratory central pattern generator termed fast synchronous oscillations, segregating into high, medium, and low-frequency bands (Ghali and Marchenko, 2013; Marchenko and Rogers, 2006a,b, 2007b, 2009; Marchenko et al., 2012).
The preBötzC was identified as the rostralmost structure requiring preservation to maintain the generation of a respiratory rhythm exhibiting monophasic inspiratory discharge, elegantly demonstrated by the seminal work of Emeritus Professor Dr. J.C. Smith et al. (1991) in the in vitro preparation of the neonatal rat. PreBötzC is located 1.8 to 2.1 mm lateral to the midline at a depth of 550 to 850 μm from the ventral medullary surface, spanning 400 μm in the rostrocaudal dimension, located 800 to 1200 μm caudal to the caudal pole of the facial nucleus and 1600 to 2000 μm rostral to the calamus scriptorius. The preBötzC contains preinspiratory (pre-I) and preinspiratory inspiratory (pre-I/I) phase spanning units with intrinsic bursting properties (Morgado-Valle and Beltran-Parrazal, 2017) and represents the principal kernel of inspiratory rhythm generation (Smith et al., 1991; Marchenko et al., 2016). The preBötzinger complex is comprised of a group of synaptically and electrotonically coupled glutamatergic (Gray et al., 2010), GABAergic (Kuwana et al., 2006), and glycinergic spontaneously depolarizing units (Morgado-Valle et al., 2010; Winter et al., 2009) exhibiting pacemaker properties utilizing persistent sodium INaP (Koizumi and Smith, 2008; Toporikova and Butera, 2011) and calcium-activated nonselective cationic currents (ICAN) (Morgado-Valle and Beltran-Parrazal, 2017; Toporikova and Butera, 2011). The spontaneous bursting activity of preBötzC units is thus driven by INaP and ICAN (Koizumi et al., 2018; Picardo et al., 2019). The source of calcium providing the activating stimulus to the ICAN current appears to differentially modify rhythm parameters, with synaptic extracellular derived calcium modifying amplitude and intracellular derived calcium modifying frequency (Phillips et al., 2019). Treatment of reduced preparations with cadmium (ICAN channel inhibitor) reduces rhythm amplitude, with riluzole (INaP channel inhibitor) completely abolishing the respiratory rhythm.
Since identification of the preBötzC as the principal inspiratory rhythm generating kernel, authors have made ignominious and valiant strides and efforts to neurochemically and neurogenetically characterize more localized and specific clusters within this nucleus to principally generate the respiratory rhythm (Gray et al., 2010; Morgado-Valle and Beltran-Parrazal, 2017; Vann et al., 2016). The identification of somatostatin receptor and Dbx1 transcription factor expressivity in preBötzC neurons initially proved an exciting and pivotal discovery, putatively identifying a specific group of units within the preBötzC as principally mediating respiratory rhythmogenesis. Pharmacogenetic inhibition of somatostatin expressing preBötzC units (Tan et al., 2008) and genetic ablation of Dbx1 expressing units (Wang et al., 2014) abolished respiratory rhythm generation and pattern formation. The BötzC contains dec post-I (Ezure and Manabe, 1988) and aug late-E (Jiang and Lipski, 1990) neurons providing powerful inhibition to preBötzC pre-I units and the rVRG aug-I cell population (Yang and Feldman, 2018) and synaptically driving the expiratory premotoneurons of the cVRG (Ezure et al., 2003; Long and Duffin, 1986; See et al., 1987; Shen et al., 2003; Tonkovic-Capin et al., 2003). The BötzC is located 1.9 to 2.2 mm lateral to the midline, at a depth of 450 to 750 μm from the ventral medullary surface, spanning a rostrocaudal dimension of 600 to 700 μm, located 100 to 750 μm from the caudal pole of the facial nucleus and 2000 to 2750 μm rostral to the calamus scriptorius (Marchenko et al., 2016).
The spontaneous bursting properties of preBötzC pre-I units (Morgado-Valle and Beltran-Parrazal, 2017) coupled with persistence of rhythmic discharge in situ and in vitro in preparations containing preBötzC though lacking the full complement of network elements and in the presence of GABAAergic and glycinergic antagonists (Smith et al., 1991; Smith and Feldman, 1987; Holtman and King, 1988; Schmid et al., 1991) was collectively interpreted by early pioneers in the field of respiratory neurophysiology to signify pacemaker mechanisms to be wholly sufficient in generating the respiratory rhythm and pattern (Fig. 3). In vitro studies carried out since have continued to variably and alternately evidence respiratory rhythm independence (Paton and Richter, 1995) or powerful modulation in vitro (Murakoshi et al., 1985; Smith and Feldman, 1987) and in vivo (Holtman and King, 1988; Schmid et al., 1991) by fast inhibitory synaptic neurotransduction. The works of Richter et al. (1986) demonstrated tonic discharge with the abolition of phasic bursting in phrenic and hypoglossal motor outputs in low chloride solutions. These results would seem to evidence a requirement of chloride-based signaling in generating the inhibitory neurotransmission requisite to generate respiratory rhythmicity. In contrast, the persistence of neural respiratory discharge following treatment with broad-spectrum antagonists of GABAAergic and glycinergic signaling provided evidence to the contrary, indicating rhythm independence of inhibitory neurotransmission (Brockhaus and Ballanyi, 1998).
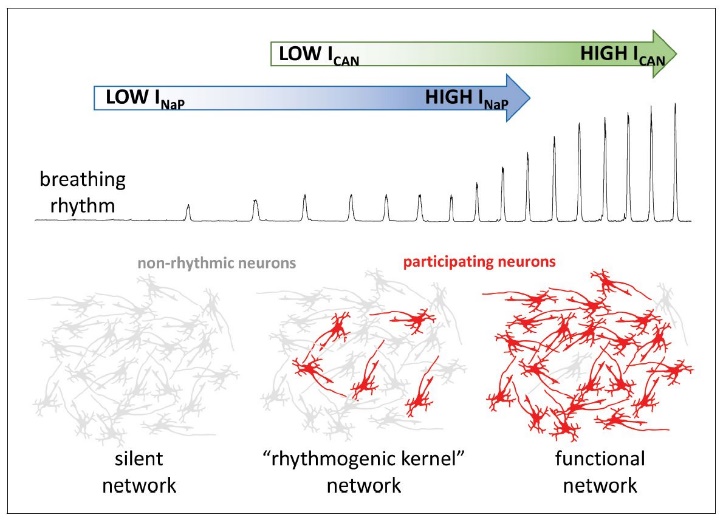 Figure 3.
Figure 3.Influences of neuronal biophysical membrane properties upon breathing rhythm. The respiratory rhythm is generated by the activities of the persistent sodium (blue) and calcium activated nonselective cationic (green) currents. Without persistent sodium channel current, the network remains silent, with neurons demonstrating nonrhythmic activity (gray). However, a few neurons with high levels of persistent sodium current may generate a weak respiratory phasic discharge, even without pacemaker cationic current. In turn, the model suggests calcium ions activate the calcium activated nonselective cationic current deriving from synaptic actions potentials. The current would then amplify interneuronal excitatory interactions. The amplificiation leads to recruitment of additional neurons participating in rhythm generation (red), producing a robust functional network. Inclusion of inhibitory elements provides the network with the emergent properties necessay to generate triphasic eupnea with inspiration and segmented expiration. Modified with permission from Fig. 1 of Ramirez and Baertsch (2019).
There exist several variations of the pacemaker model of breathing. According to the simplest construct, the spontaneous discharge of preBötzC pre-I units generates the breathing rhythm (Ramirez and Baertsch, 2019; Smith et al., 1991). Onimaru and colleagues (Onimaru et al., 1987, 1988, 2003, 2006; Onimaru and Homma, 2006) have strongly advocated and advanced the concept of RTN/pFRG spontaneously bursting units principally originating the pre-I drive conveyed to preBötzC units. The RTN consists of a group of exquisitely chemosensitive units expressing the transcription factor Phox2b, mediating central chemoreception, and providing the diffuse excitatory drive to medullary ventral respiratory column nuclei and dorsal respiratory group, extensively characterized by the works of Guyenet et al. (2019). Specifically, a cluster of neurons located more laterally in pFRG appears to exhibit oscillatory properties exhibiting pre-I and expiratory biphasic activity and may drive the preBötzC pre-I units (Anderson and Ramirez, 2017; Guyenet et al., 2019). These properties were alternatively interpreted to indicate the existence of an inspiratory expiratory oscillator, with the preBötzC representing the kernel of inspiratory rhythm generation and the RTN/pFRG representing the soi-disant locus of expiratory rhythm generation (Janczewski and Feldman, 2006). In very early neonates, opioids abolish the rhythmic discharge of preBötzC pre-I units, though RTN/pFRG neurons continue to burst spontaneously, apparently evidencing opioid resistance (Ballanyi et al., 2009; Mellen et al., 2003; Onimaru et al., 2006). Authors have thus suggested the RTN/pFRG to be utilized in the immediate period following birth, given inhibition of the preBötzC oscillator by the parturition induced opioid surge.
We shall thus make our best efforts to discourse upon the nature of the preBötzC pacemaker cells (Gray et al., 2010; Rybak et al., 2014). The oscillators drive the network mechanism natively and may support monophasic bursting in the presence of compromised inhibitory network elements (physical network reduction, antagonism of synaptic inhibition, severe hypoxia). The preBötzC is comprised of pre-I units exhibiting spontaneously bursting activity according to the biophysical properties of persistent sodium and calcium-activated nonselective cationic currents. PreBötzC intrinsic bursting units may utilize either or both of these currents (Toporikova and Butera, 2011). These pacemaker currents become refractory consequent to inactivation of the channels proper and blunting of the electrogenic net outwardly electropositive adenosine-triphosphatase dependent sodium-potassium exchange consequent to intracellular accumulation of sodium (Rybak et al., 2003b, 2014; Toporikova and Butera, 2011; Wu et al., 2005). Specifically, the persistent sodium current undergoes inactivation and the calcium activated cationic currents undergo Ca2+-dependent inositol triphosphate (IP3) receptor inactivation. The activation and inactivation kinetics of persistent sodium and calcium activated nonselective cationic currents thus contribute prominently to determining rhythmic bursting properties of the preBötzC (Toporikova and Butera, 2011). The persistent sodium channel inactivation mechanism exhibits rapid kinetics, with time to onset time constant (τonset) of 2.04 seconds and time to recovery time constant (τrecovery) of 2.21 seconds, demonstrated by Wu and colleagues (Wu et al., 2005) in neurons of the mesencephalic trigeminal nucleus.
Gradually augmenting the tonic drive of a cell from 0 to 100% confers rhythmic activity upon inactive units, eventually transitioning and transforming into tonic discharge. The bifactorial influence of tonic drive and cationic (INaP or ICAN) conductance thus multivariably influences whether a given unit operates in bursting mode, discharges tonically, or remains inactive. According to computational modeling simulations, units exhibiting dual persistent cationic currents and those with dual mechanistic persistent cationic current inactivation exhibit a broader dynamic range of rhythmic bursting compared to those utilizing individual persistent cationic currents or monomechanistic inactivation. The physiological range of rhythmic bursting is also broader in units with a more complex biomolecular architecture of spontaneously depolarizing currents. Thus, varying the tonic drive in pacemaker cells with several spontaneously depolarizing currents exhibiting multiple mechanisms of inactivation smoothly modifies unit discharge frequency. In contrast, varying the tonic drive in pacemaker cells with individual spontaneously depolarizing currents, exhibiting simple mechanisms of inactivation, may cause abrupt transitions from inactivity to tonic discharge.
Tonic conductance critically determines burst duration. Decreasing INaP conductance to 0 causes the rhythmic bursting frequency to become exquisitely sensitive to slight changes in the tonic drive. Modeling units with variable calcium-activated nonselective cationic current (INaP = 0) generates substantive bursts. Modeling units with variable persistent sodium conductance and calcium-activated nonselective cationic current (ICAN = 0) generates short-duration bursts. These results appear at apparent conflict with the experimental evidence derived in vitro by Pena and colleagues (Peña et al., 2004). Combined antagonism of synaptic inhibitory neurotransmission was utilized to selectively reveal cadmium-insensitive (INaP) and cadmium-sensitive (ICAN) pacemakers. Cadmium insensitive pacemaker bursting was transformed into tonic discharge by treatment with riluzole and cadmium sensitive pacemaker bursting was completely abolished by cadmium treatment. Thus, the multiplicative interaction of the level of tonic drive and synaptic weighting exhibits heterogeneity across cells utilizing individual versus multiple spontaneously depolarizing currents. Pacemaker cells with greater spontaneously depolarizing currents constituting the intrinsic bursting mechanism exhibit rhythmic bursting across varying levels of tonic drive and synaptic activity (Fig. 4). At a given synaptic weighting, the pacemaker cell maintains rhythmic bursting across a broader range of tonic drive. Reciprocally, at a given tonic drive, the pacemaker cells maintain rhythmic bursting across a broader range of synaptic weighting.
 Figure 4.
Figure 4.Persistent sodium dependent bursting occurs with low intermediate levels of tonic drive across a dynamic range of synaptic weighting. Combined INaP and ICAN-dependent bursting occurs with intermediate levels of tonic drive across a slightly greater dynamic range of synaptic weighting. INaP and ICAN-independent bursting occurs across a wide range of tonic conductance and synaptic weighting. In the presence of high tonic drive, ICAN-dependent bursting occurs across a narrower range of synaptic weighting. At low or high extremes of tonic conductance or synaptic weighting, pacemaker units are alternatively inactive or exhibit tonic discharge. Modified with permission from Fig. 6 of Rybak et al. (2014).
Models of respiratory rhythmogenesis and pattern formation are predicated upon fundamental neurophysiological principles and sets of differential equations. The works of Rybak and colleagues (2014) and Molkov et al. (2017) have largely provided us with our current computational conceptualization of respiratory central pattern generation. The directionality of a membrane ion current is determined principally by the net Nernst electrochemical potential acting upon a variable ion, Z:
Membrane potassium leak ionic conductance is derived from Nernst electrochemical potentials and relative sodium and potassium membrane permeabilities:
where pNa/K is the relative membrane permeability of sodium and potassium ions, [Na+]o and [Na+]i and [K+]o and [K+]i are the extracellular and intracellular concentrations of sodium and potassium, respectively. Resting membrane voltage is determined by the integrated effects of the net electrochemical driving forces acting upon each ion according to the Goldmann-Katz equation:
where R (8.314462 Joules per mole per degree Kelvin) represents the gas constant utilized in the physical chemistry equation PV = nRT, T represents temperature (in Kelvin), z is the ionic charge, and F is Faraday’s constant (electrons per mole of ion; coulombs per mol). The opening of chloride ion channels alternatively generates membrane voltage hyperpolarization and/or shunting inhibition, according to the initial resting membrane potential (Loria et al., 2013). Computational models of individual neuronal and population activity may be generated incorporating various membrane channels and ion currents, including fast sodium, persistent sodium, calcium-activated nonselective cationic, delayed rectifier potassium, leak potassium, general excitatory synaptic, and general inhibitory synaptic current according to Hodgkin Huxley formalism. The molecular biophysics of these channels may alternatively be modeled utilizing maximal or constant conductance. Constants and relations are derived from empirical data. The models, in turn, are tested according to their ability to predict novel behavior at the network level in experiments. The total membrane ionic current $∑_{m=1}^{g}(I_m)$ is proportional to the product of the total capacitance, C, and the derivative of voltage with respect to time, dV/dt:
C dV/dt=$∑\limits_{m=1}^{g}(I_m)$
Let us model the simplest of synaptic interactions. The conductance of any neuron within the network g reflects the sum of the extra network (gEdr) and network (gNdr) excitatory synaptic drive:
Population models are generated by the emergent integration of the computational properties of individual neuronal interactions. A given neuron projects to neuron b and delivers a spike at the time tab, increasing the channel conductance by a constant GZ per synaptic input wab, with the total increase in the cellular conductance proportional to the product of the monospike incremental increase in conductance and synaptic weighting, GZwab. Dynamics of neuronal conductance in the efferent neuron b at any given time, t, is accordingly represented by the following formalism.
$G_{netb}(t)=G^Z∑\limits_{a(a≠1)}w^{ab}∑exp\frac{-[t-t^{ab}]}{[T_synE]}$
where Gnetb is the net membrane conductance of neuron b, GZ is the incremental increase in membrane conductance in response to the arrival of one action potential spike from neuron a to neuron b. tab represents the time at which the spike arrives, wab represents the synaptic weighting, and TsynE represents the time constant of excitatory synaptic inputs. The proportionality t-tab represents the difference in time between efferent neuron synaptic conductance and spike arrival. These models generate bursting behavior with properties and multivariate interactions powerfully modulable and inferable by the elegant interaction of the equations and mathematical formalism.
The presented confluence of findings has presented quite the empirocotheoretical conundrum and inspired fruitful, though occasionally nonmaliciously heated, debate (Dutschmann and Herbert, 2006; Feldman and Janczewski, 2006a,b; Onimaru et al., 2006). In this regard, investigators have extensively debated the role of fast inhibitory synaptic transmission in respiratory rhythmogenesis and pattern formation (Dutschmann and Paton, 2002; Marchenko et al., 2016; Molkov et al., 2017; Smith et al., 2007). While the role of GABAAergic and glycinergic signaling in respiratory rhythmogenesis and pattern formation has represented the crux and pivotal point at the core of this debate (Marchenko et al., 2016), further studies are clearly necessary in order to provide us with an equivalently in-depth understanding of the GABABergic contribution to modulation of respiratory rhythmogenesis and pattern formation in adults, juveniles, and neonates (Pierrefiche et al., 1993). We suggest a modulatory role of GABABergic signaling in adults and a more critical role in neonates to breathing generation.
The spontaneously bursting oscillators of the neural respiratory circuitry (Bonis et al., 2010; Ghali and Marchenko, 2016a; Gray et al., 2010; Malheiros Lima et al., 2018; Morgado-Valle and Beltran-Parrazal, 2017; Onimaru and Dutschmann, 2012) are incorporated within an elegantly complex though empirically dissectable network utilizing mutual and reciprocal excitatory and inhibitory synaptic interactions to generate the breathing rhythm (Molkov et al., 2017). Powerful modulation or alteration of the respiratory pattern by systemic administration or local pressure microinjections of pharmacological agonista and antagonists of GABAAergic, glycinergic, and GABABergic signaling in vivo would seem to provide substantive evidence validating a critical role of both fast inhibitory and slow neuromodulatory synaptic transmission in shaping the breathing pattern (Brockhaus and Ballanyi, 1998; Haji et al., 1990; Johnson et al., 2002; Pfeiffer and Zhang, 2007; Pierrefiche et al., 1993; Zhang et al., 1999, 2002;). The pioneering work of Marchenko et al. (2016) demonstrated significant respiratory rhythm perturbations following microinjections of the GABAAergic antagonist gabazine and glycinergic antagonist strychnine in BötzC or preBötzC in anesthetized rats and unanesthetized decerebrate juvenile rats, signifying a critical role of fast inhibitory neurotransmission in breathing generation (Marchenko et al., 2016).
We thus present, detail, and expand upon a network model of respiratory rhythmogenesis and pattern formation predicated upon the intact functionality of inhibitory network elements according to the model of Molkov and colleagues (Molkov et al., 2017) (Fig. 5). Oscillators disparately distributed throughout the brainstem (Anderson and Ramirez, 2017; Anderson et al., 2016; Guyenet et al., 2019) interact with and modulate the discharge of, preBötzC spontaneously bursting units exhibiting pre-I activity (Malheiros Lima et al., 2018; Morgado-Valle and Beltran-Parrazal, 2017) driving the rhythmic discharge of rVRG aug-I neurons (Molkov et al., 2017; Okazaki et al., 2001), while inhibiting BötzC dec post-I and aug late-E units (Molkov et al., 2017; Richter et al., 1986; Schwarzacher et al., 1991). PreBötzC dec early-I units (Morgado-Valle and Beltran-Parrazal, 2017) provide inhibition to BötzC dec post-I (Ezure and Manabe, 1988) and aug late-E (Jiang and Lipski, 1990) units and rVRG aug-I neurons (Rybak et al., 2014), generating the augmenting spatiotemporal dynamics of inspiratory discharge manifest in brainstem and spinal motor outputs (Molkov et al., 2017; Schmid et al., 1996).
 Figure 5.
Figure 5.Closed-loop model of neural respiratory network. The respiratory rhythm is generated by the activity of preBötzinger complex spontaneously bursting preinspiratory and preinspiratory inspiratory phase spanning units. These units drive the discharge of rostral ventral repsiratory group inspiratory neuronal discharge and shape the augmenting pattern of the spatiotemporal dynamics. PreBötzinger complex decrementing early inspiratory neurons provide inhibition to rostral ventral respiratory group units and prominently contribute to shaping the augmenting pattern of discharge. These preBötzinger complex units receive prominent inhibition by Bötzinger decrementing postinspiratory and augmenting late expiratory units. Bötzinger decrementing post-inspiratory units derive tonic excitatory inputs from Kölliker-Fuse nucleus and parafacial respiratory group located in the dorsolateral metencephalic tegmentum. Bötzinger complex decrementing postinspiratory and augmenting late expiratory units exhibit mutual and reciprocal inhibitory interactions. These units provide phasic excitatory drive to caudal ventral respiratory group premotoneurons supplying abdominal expiratory motoneurons. Spontaneously bursting parafacial respiratory group pre-inspiratory group oscillators drive the discharge of preBötzinger complex pre-inspiratory units and drive hypercapnia induced active expiration. Rostral ventral respiratory group augmenting inspiratory and late inspiratory units provide premotoneuronal drive to inspiratory motoneurons in hypoglossal, phrenic, and external intercostal motor nuclei. Caudal ventral respiratory group premotoneurons drive the discharge of brainstem upper airway related and spinal cord internal intercostal and abdominal expiratory motoneurons. Abd., abdominal; AbN, abdominal nerve; aug-E, augmenting expiratory neuron; BötC, Bötzinger complex; CPG, central pattern generator; cVRG, caudal ventral respiratory group; early-I, early-inspiratory neuron; late-E, late-expiratory neuron; Mns, motoneurons; NTS, nucleus of the tractus solitarius; P-cells, Pump cells; P(e), excitatory pump cells; P(i), inhibitory pump cells; pFRG, parafacial respiratory group; PN, phrenic nerve; post-I, post-inspiratory neuron; pre-BötC, pre-Bötzinger complex; pre-I/I, pre-inspiratory/inspiratory phase-spanning neuron; PSRs, pulmonary stretch receptors; ramp-I, ramp-inspiratory neuron; RTN, retrotrapezoid nucleus; rVRG, rostral ventral respiratory group; VRC, ventral respiratory column. Modified with permission from Fig. 6 of Molkov et al. (2017).
The BötzC contains dec post-I units of principally glycinergic and aug late-E units of principally GABAergic character (Champagnat et al., 1982; Ezure and Manabe, 1988; Haji et al., 1992; Molkov et al., 2017; Richter et al., 1986, 1987; Schmid et al., 1996; Schwarzacher et al., 1991). BötzC dec post-I units (Ezure and Manabe, 1988) provide prominent inhibition to preBötzC pre-I, pre-I/I phase-spanning, and dec early-I units (Morgado-Valle and Beltran-Parrazal, 2017) and rVRG aug-I neurons (Molkov et al., 2017). The sequential activity of medullary late-I units and BötzC dec post-I units in conjunction with that of effectively generates inspiratory off switching late-I (Smith et al., 2007, 2009; Rybak et al., 2014; Marchenko et al., 2016). BötzC aug late-E units (Jiang and Lipski, 1990) provide prominent inhibition to preBötzC pre-I and dec early-I propriobulbar interneurons and rVRG aug-I premotoneurons (Jiang et al., 1990; Molkov et al., 2017; Rybak et al., 2014), which provide monosynaptic and polysynaptic premotoneuronal inputs to cranial and spinal motor nuclei (Ellenberger and Feldman, 1990; Marchenko et al., 2016; Smith et al., 2007; Stornetta et al., 2003). BötzC dec post-I (Ezure and Manabe, 1988) and aug late-E (Jiang and Lipski, 1990) units exhibit mutual inhibitory interactions and their discharge exhibits mutual inhibitory interactions (Molkov et al., 2017; Rybak et al., 2014). Thus, the neuroanatomic circuitry and mechanism of GABAAergic and glycinergic signaling becomes readily evident from the described model of respiratory rhythmogenesis and pattern formation (Marchenko et al., 2016; Molkov et al., 2017). These inhibitory mechanisms critically require the integrity and functionality of inhibitory mechanisms utilizing fast chloride conductances (Marchenko et al., 2016).
Thus, in brief, BötzC glycinergic dec post-I neurons (Ezure and Manabe, 1988; Richter et al., 1986, 1987; Schwarzacher et al., 1991) provide inhibition to all neurons in the ventral respiratory column nuclei and, contemporaneously with late inspiratory neurons (Cohen et al., 1993), mediate inspiratory off switching (Bianchi et al., 1995; Marchenko et al., 2016; Molkov et al., 2017; Richter, 1996; Rybak et al., 2014; Smith et al., 2007, 2009; Okazaki et al., 2001; Yamazaki et al., 2000). GABAergic BötzC aug late-E neurons (Jiang and Lipski, 1990) inhibit BötzC dec post-I neurons (Ezure and Manabe, 1988), as well as inspiratory neurons (Rybak et al., 2014). PreBötzC pre-I neurons drive the discharge of preBötzC dec early-I neurons, which provide inhibition to BötzC expiratory units and rVRG aug-I neurons (Cui et al., 2016; Stornetta et al., 2003; Tryba et al., 2006; Viemari et al., 2011). This network model of breathing may be used to accurately explain the findings of various investigations reducing network connectivity mechanotransectively or via antagonism of synaptic inhibition. Pontomedullary transectiontransforms triphasic eupnea to a two-phase rhythm consisting of neural inspiration and expiration (Molkov et al., 2017; Smith et al., 2007, 2009). This likely results from the removal of excitatory tonic drive provided to BötzC glycinergic dec post-I units by Kölliker-Fuse and medial parabrachial nuclei and to BötzC GABAergic aug late-E units by the RTN (Molkov et al., 2017; Smith et al., 2007, 2009). The use of low chloride perfusates similarly reduces triphasic eupnea, with neuronal depolarization occurring consequent to the loss of synaptic inhibition (Richter et al., 1986). The two-phase neural respiratory rhythm manifests peripherally as loss of cervical vagus post-I activity and transformation of the vagal, hypoglossal, phrenic, and intercostal neurogram aug-I burst shape to bell-shaped patterns. In respiratory-related nuclei, the two-phase rhythm manifests as decrementing expiratory and inspiratory activities in BötzC and preBötzC neuronal discharge. Pontomedullary transection amplifies rhythmic respiratory variability and decreases amplitude, presumably by excluding metencephalic excitatory and inhibitory influences promoting synchrony amongst the myelencephalic core rhythm generating elements. These effects generate short duration ectopic bursting intermixed with longer duration square wave bursts. In preparations generating the two-phase rhythm, riluzole effectively eliminates ectopic bursting (Smith et al., 2007, 2009).
Transection between the BötzC and preBötzC reduces two-phase myelencephalic respiratory rhythmicity to monophasic decrementing bursting (Smith et al., 2007, 2009). Absent reciprocal mutual inhibitory interactions between the BötzC and preBötzC (Smith et al., 2007), the preBötzC operates in pacemaker mode via the regular and rhythmic discharge of spontaneously bursting pre-I and pre-I/I units (Morgado-Valle and Beltran-Parrazal, 2017). This pattern of monophasic decrementing inspiratory bursting bears a striking resemblance to gasping in rhombomyelic intact models and high cervically transected unanesthetized precollicular decerebrate rats (Ghali and Marchenko, 2016a). In BötzC/preBötzC transected preparations, persistent sodium channel current antagonism with the agent riluzole progressively reduces neural respiratory frequency and amplitude, eventually abolishing the rhythm, not restorable by hypoxic stimulation, hypercapnia, extracellular hyperkalemia, or any combination thereof. Transection between the preBötzinger complex and the rostral ventral respiratory group eliminates all neural respiratory rhythmic activity, not restorable with chemosensory stimulation. Blockade of preBötzC persistent sodium channels utilizing riluzole thus abolishes respiratory rhythm in slice preparations and in situ and in vivo preparations following transection between the BötzC and preBötzC (Koizumi and Smith, 2008; Paton et al., 2006; Rybak et al., 2003), but not in vivo or in situ preparations with preserved Bötzinger complex and/or pontine connectivity with preBötzC, since in these intact preparations, preBötzC continues to receive tonic and phasic excitatory and inhibitory network inputs, obviating the need for intrinsic bursting pacemaker cells (Molkov et al., 2017; Smith et al., 2007, 2009).
In order to develop a thorough understanding of the effect of GABA upon the brainstem neural respiratory circuitry, it will first prove prudent to discourse upon the structure and function of the fundamental biomolecular machinery mediating the described modulations (Martínez-Campos et al., 2019; Sadeghi et al., 2018). We shall have as our principal purpose to underscore and highlight the significance of the critical features of GABAAergic and GABABergic signaling as they contribute to generating and modulating the breathing rhythm and pattern (Bowery et al., 1990; Connors, 1992; Cramer et al., 2010; Gonzalez Burgos, 2010; Kohl and Paulsen, 2010; Sil'kis, 1996). These effects are well demonstrated and detailed in appropriately designed studies (Brockhaus and Ballanyi, 1998; Haji et al., 1990; Johnson et al., 2002; Pierrefiche et al., 1993; Zhang et al., 2002), though neither widely appreciated nor incorporated in models of respiratory rhythmogenesis and pattern formation (Molkov et al., 2014; Rybak et al., 2014). A critical synthesis of the major findings of these studies thus proves prudent in order to provide a more robust and complete mechanistic theoretical basis forming and contributing to a thorough conceptualization of the full complement of the fundamentals of inhibitory neurotransmission in neural respiratory circuitry and networks inclusive of inhibitory elements in general (Dutar and Nicoll, 1988; Gahwiler and Brown, 1985; Malitschek et al., 1998; Misgeld et al., 1995; Pierrefiche et al., 1993; Pfeiffer and Zhang, 2007; Wagner and Dekin, 1993, 1997; Zhang et al., 1999, 2000).
The amino acid glycine and the amino acid derivative gamma-aminobutyric acid (GABA) represent the principal neurochemical mediators generating fast (Dogas et al., 1998; Fregosi et al., 2004; Pierrefiche et al., 2004; Ritter and Zhang, 2000; Schmid et al., 1991, 1996; Vedyasova and Kovaleva, 2018) and slow (Lebedeva et al., 2010; Pfeiffer and Zhang, 2007; Pierrefiche et al., 1993; Schmid et al., 1989; Wagner and Dekin, 1993, 1997) inhibitory synaptic neurotransmission in neural respiratory circuitry of the brainstem and spinal cord. Fast inhibitory synaptic mechanisms constitute those pathways mediated by the opening of receptor linked ion channels, generating net inward fluxes of chloride anion (Kardos, 1993; Majewska, 1990; Tanelian et al., 1993; Vicini, 1991; Xu et al., 2019). Slow neuromodulatory inhibition comprises those mechanisms activated through intracellular signal transduction pathways by receptor-ligand binding and mediating the opening or closing or modifying the molecular biophysical properties of membrane ion channels. The activated currents transmit net outward potassium conductances (Breton and Stuart, 2017; Pfeiffer and Zhang, 2007; Wagner and Dekin, 1993) and the inhibited currents transmit net inward calcium conductances in electroconductive neurolemmal membranes (Cai et al., 2018; Gahwiler and Brown, 1985; Li and Guyenet, 1995; Misgeld et al., 1995; Mizuta et al., 2008; Newberry and Nicoll, 1985; Nicoll et al., 1990; Thalmann, 1988; Zhang et al., 1999).
The neuromodulator gamma-aminobutyric acid is synthesized from the precursor acidic amino acid glutamate, constituted by a core carbon atom tetravalently bonded to a hydrogen atom, amino group, carboxyl group, and ionized carboxylate alkyl side chain moieties through sp3 hybridized orbitals, by the catalytic activity of the enzyme glutamate decarboxylase, of which there exists isoforms of 65 and 67 kDa molecular weight, a reaction which occurs in synaptic terminals (Ueno, 2015). Synaptovesicular GABA transporters mediate the uptake and loading of the GABA neurotransmitter into synaptic vesicles (Santos et al., 2013). GABA loaded synaptic vesicles subsequently undergo fusion with the presynaptic membrane upon arrival of an action potential transmitted the length of the axon, generating voltage mediated activation of voltage-gated calcium channels (see Rosa and Fratangeli, 2010 for an excellent review). Glycine is a nonpolar amino acid with a core carbon atom covalently tetravalently bonded to two hydrogen atoms, an amino group, and a carboxyl group through sp3 hybridized orbitals, lacking an alkyl side chain. Some presynaptic terminals corelease GABA and glycine. The functional implications of these heterogeneous inhibitory synapses remain to be determined, though they may play a critical role in organizing inhibitory network mechanisms in neural respiratory circuitry.
GABAA (Scott and Aricescu, 2019) and GABAB (Tomita, 2019) receptors constitute the two principal general subtypes of neurolemmal γ-aminobutyric acid receptors, comprising a group of evolutionarily conserved heterodimeric transmembrane receptors with amino-terminal extracellular ligand binding and carboxyl-terminal intracellular effector domains. The differential effects of GABA exerted upon presynaptic, postsynaptic, or extrasynaptic zones is thus principally mediated by the differential and alternate locoregional heterogeneity of synaptodendritic, somatic, and extrasynaptic cell surface expression of different types of GABA receptor subtypes, contemporaneously and alternatively augmenting or attenuating cationic and anionic conductances in the neurolemmal membrane (Gahwiler and Brown, 1985; Misgeld et al., 1995; Newberry and Nicoll, 1985; Nicoll et al., 1990; Thalmann, 1988).
The GABAA receptor represents an evolutionarily conserved heteropentameric receptor consisting of α-helices and β-pleated sheets with multiple ligand binding zones (Martínez-Campos et al., 2019). GABA ligand or synthetic agonist binding to the GABAA receptor generates immediate conformational changes of the receptor effecting the opening of an anion channel transmitting a selective chloride-based conductance (Fig. 6) (Kardos, 1993; Majewska, 1990; Tanelian et al., 1993; Vicini, 1991; Xu et al., 2019). Nernst electrochemical potentials acting upon the ion determine the directionality of net ionic flux across the neurolemmal membrane. The opening of chloride ion channels alternatively generates hyperpolarization of the membrane voltage and/or shunting inhibition, according to the initial resting membrane potential (Loria et al., 2013). Benzodiazepines variably augment the effects of GABAergic neurotransmission by amplifying the frequency of channel opening, with barbiturates potentiating GABAergic signaling by augmenting duration of channel opening.
 Figure 6.
Figure 6.GABAA heteropentameric γ-aminobutyric acid type A receptors (GABAARs). A: Cryoelectron microscopic determination of overall and heteropentameric structure of GABAA receptor. B: GABAA receptor architecture demonstrated viewed from the extracellular space with the receptor subunit configuration depicted schematically with glycans presented in in ball and stick representation. C: GABA receptor architecture viewed from the cytosolic side looking into the ion-conducting pore (bottom view). D: Diagramatic demonstration of extracellular receptor glycosylation sites with glycans and critical amino acid residues mediating their binding demonstrated in ball and stick representation. E: Schematic representation of the underlying mechanism governing synaptic heteropentameric GABAA receptor assembly. The scheme demonstrates glycosylation of the conserved asparagine 111 (Asn111) residue importantly contributes a critical structural role in receptor assembly, which also determines the order of subunit arrangement. (F-L) GABAA receptor structures bound to various ligands. (F) The heteropentameric GABAA receptor is depicted in cartoon representation along with structurally validated ligands in the space-filling representation. Enlarged views demonstrate GABAA receptor binding pockets of the naturally biologically generated agonist GABA (PDB: 6HUJ, G), the positive allosteric modulator (PAM) diazepam (PDB: 6HUP, H-I), the competitive receptor antagonist bicuculline (PDB: 6HUK, J), the receptor channel pore blocker picrotoxin (PDB: 6HUG, K) and the lipid phosphatidylinositol-2 (PDB: 6I53, L). Enlarged views of specific GABAA receptor structural components reflect the color of the box indicated in the overall GABAA receptor structure displayed in (F). In panels F-L, all ligands and critical residues mediating binding are demonstrated in ball and stick models and the protein chains are demonstrated schematically. Modified with permission from Fig. 1 of Kasaragod and Schindelin (2019).
The GABAB receptor is a heterodimeric G protein-coupled receptor (White et al., 1998; Waldvogel et al., 2004) comprised of an R1 subunit, with an extracellular N terminal ligand-binding domain, and an R2 subunit, containing an intracellular effector coupling domain (Kasaragod and Schindelin, 2019) (Fig. 7). Expression of differential splice variants of the GABAB receptor could putatively generate differential kinetics of signaling mediated by ligand binding through distal intracellular signal transduction effects (Fritschy et al., 1999). The GABAB receptor demonstrates wide expressivity throughout the cerebrum (Connors, 1992; Kuriyama et al., 1993; Malcangio and Bowery, 1995; Sil'kis, 1996), brainstem (Burman et al., 2003; Jurčić et al., 2019; Pfeiffer and Zhang, 2007;), cerebellum (Kuriyama et al., 1993; Malcangio and Bowery, 1995), and spinal cord (Lev-Tov et al., 1990; Malcangio and Bowery, 1995), as well as cells of a plethora of peripheral effect organs (Castelli et al., 1999), including pancreatic beta islet cells (Braun et al., 2004; Crivello et al., 2013), adrenocortical cells (Häusler et al., 1993; Metzeler et al., 2004), cardiomyocytes (Lorente et al., 2000), chondrocytes (Tamayama et al., 2005), and osteoblasts (Fujimori et al., 2002). Studies have variably elucidated the functional significance of these peripheral GABAB receptors in generating and modulating the breathing rhythm and neural circuitry underlying the genesis of various rhythmic and patterned behavioral outputs.
 Figure 7.
Figure 7.GABAB receptor ligand binding zone. The GABAB receptor is a transmembrane heterodimeric G protein coupled receptor comprised of R1 and R2 with an N-terminal ligand binding zone and a C-terminal effector coupling domain. Activation of the GABAB receptor variably activates outwardly directed potassium and/or inhibits inwardly directed calcium conductances on the presynaptic and/or postsynaptic membranes through signal transduction pathways. GABAB receptor activation mediates slow inhibitory synaptic neuromodulation, requiring intermediate biochemical reactions and signal transduction mechanisms to effect a change in membrane channel biokinetics and voltage dynamics. This is contrasted with the heteropentameric GABAA receptor, ligand binding to which generates and immediately activates a chloride conductance, mediating fast inhibitory synaptic neurotransmission. Both types of GABA receptors have a myriad of natural and synthetic ligand binding zones. This biomolecular diversity provides a mechanism putatively generating a range of heterogeneity of structural functional coupling. Amino acids (colored thin sticks) constituting the GABAB active binding site and (R)-baclofen (green sticks) are schematically depicted. Modified with permission from Fig. 3 of Martínez-Campos et al. (2019).
GABAA receptors characteristically localize postsynaptically on dendrites and cell somata, though they may also be found extrasynaptically. GABAB receptors are diffusely distributed synaptodendritically, somatically, and axonally, localizing to presynaptic terminals (Delaney and Crane, 2016; Orts-Del'Immagine and Pugh, 2018), postsynaptic membranes (Chalifoux and Carter, 2011; Crunelli and Leresche, 1991; Gonzalez-Burgos, 2010; Mouginot and Gähwiler, 1996; Serrats et al., 2017), or extrasynaptic (Pham et al., 1998) zones. GABAB receptors located within synaptic zones are generally of low affinity (Carpenter et al., 2012), appropriately suited given the extremely high concentration of GABA generated by synaptovesicular fusion and release of neurotransmitter within the confines of the synaptic cleft, contrasted with the very high ligand binding affinity demonstrated by GABA receptors located in extrasynaptic zones (Barolet et al., 1985; Gonzalez Burgos, 2010). This represents a necessary and wise construct of nature conferring modulatory influence upon extrasynaptic signaling given the significantly lower amounts of GABA diffusing to these regions (personal communication, Emeritus Professor Dr. Vitaliy Marchenko). We assert this neurostructural organization is thus aptly suited and designed to mediate and convey the effects of GABA upon neurotransmission within the presynaptic, postsynaptic, and extrasynaptic zones. Oxygen tension appears to critically modulate and influence the expression patterns of the GABAB biomolecular machinery (Anju et al., 2010), with hypoxia effecting significant reductions of expression of total GABA and GABAB receptors and glutamate decarboxylase and increases of hypoxia-inducible factor-1α (Anju et al., 2010). These effects were readily reversed by coadministration of oxygen and glucose (Anju et al., 2010).
Activation of GABAA receptors amplifies chloride conductances exhibiting rapid kinetics independent of intracellular signal transduction pathway signaling(Majewska, 1990; Vicini, 1991; Xu et al., 2019). This chloride conductance is outwardly directed during late fetal and early neonatal development and inwardly directed during late neonatal development, juvenility, and adulthood (Ghali and Beshay, 2019). In contrast, GABA ligand binding to GABABergic metabotropic receptors generates a much slower activation of membrane potassium conductances and/or attenuation of calcium conductances. Activation of the GABAB metabotropic receptor amplifies potassium (Breton and Stuart, 2017; Fritzius and Bettler, 2019; Pfeiffer and Zhang, 2007; Wagner and Dekin, 1993, 1997), and attenuates calcium, ionic conductances (Cai et al., 2018; Li and Guyenet, 1995; Mizuta et al., 2008; Zhang et al., 1999), by coupling to and activating Gi protein. This mechanism effects the inhibition of adenylate cyclase catalytic activity (Pérez-Garci et al., 2012). The GABABergic modulated potassium conductances are located postsynaptically (Pfeiffer and Zhang, 2007; Wagner and Dekin, 1993, 1997) and the modulated calcium conductances localize to both the postsynaptic and presynaptic membranes. These receptor linked ion channels thus represent the principal biomolecular machinery mediating the effects of GABA upon neuronal transmission and neurochemical transduction. GABAB metabotropic receptor activation also activates and amplifies mitogen-activated protein kinase-dependent pathways (Gutkind, 1998). These pathways may be implicated in synaptic neuroplasticity of central respiratory circuitry.
GABAAergic signaling appears to play a critical, if not indispensable, role in respiratory rhythm generation and pattern formation, according to the highest quality in vivo experimental evidence (Marchenko et al., 2016). GABA ligand binding to neurolemmal GABAAergic receptors effects the immediate opening of membrane chloride ion channels transducing net outward fluxes during late fetal and early neonatal development and inward fluxes during late neonatal development, the juvenile period, and adult age (Ghali and Beshay, 2019). A role for GABABergic signaling is also well characterized in respiratory rhythm generation and pattern formation (Brockhaus and Ballanyi, 1998; Haji et al., 1990; Johnson et al., 1996; Pfeiffer and Zhang, 2007; Pierrefiche et al., 1993; Zhang et al., 2000, 2002), though has received comparatively less experimental investigation compared with fast inhibitory synaptic transmission utilizing GABAAergic and glycinergic mechanisms (Bongianni et al., 2010; Marchenko and Rogers, 2009; Marchenko et al., 2015; 2016; Paton and Richter, 1995; Ritter and Zhang, 2000). GABAB receptors are present and functional in the ventral respiratory column nuclei (Alheid and McCrimmon, 2008; Ballanyi et al., 1999; Bongianni et al., 2009, 2010; Haji et al., 2000; Hayashi and Lipski, 1992; Schmid et al., 1989; Zhang et al., 2002). The nature and properties of GABABergic signaling in the neural respiratory network of adult animals (Haji et al., 1990; Hay and Lindsley, 1995; Lipski et al., 1990; Pierrefiche et al., 1993) and neonatal preparations (Brockhaus and Ballanyi, 1998; Zhang et al., 2000, 2002) varies among and between different preparation types (Haji et al., 2000; Schmid et al., 1989; Zhang et al., 2002). Activation of GABAB receptors by natural or synthetic ligand binding induces membrane voltage hyperpolarization (Brockhaus and Ballanyi, 1998; Dutar and Nicoll, 1988; Johnson et al., 1996) by activating a cAMP-sensitive outward rectifying potassium conductance and/or inhibiting low or high voltage-activated calcium conductances, acting either presynaptically to reduce the release of neurotransmitter or postsynaptically by generating inhibitory postsynaptic potentials (IPSPs) (Bowery et al., 1980). Wagner and Dekin (1993, 1997) have demonstrated and characterized the kinetics and current-voltage relationships of these channels. Differential effects exerted upon the physiologies through the actions of GABAB agonists suggest the receptor to be a complex multimodulable structure (Canning et al., 2012).
Tonic and modulable membrane potassium conductances contribute prominently to generating the resting membrane potential (Flynn et al., 1999; Köhler et al., 1983; Steinhardt et al., 1972; Walz et al., 1984). These neuronal electrochemical properties are well characterized across several studies, since the works of Hodgkin and Huxley (1945, 1952, 1990) demonstrating a critical contribution of potassium conductances in contributing to resting membrane voltage, the repolarization phase of the action potential, and afterhyperpolarization discharge in the squid giant axon (Hodgkin and Huxley, 1990, 1945, 1952). Potassium conductances may be constitutively and tonically active, admitting the contemporaneous free influx and efflux of potassium ions with the directionality of net flux varying according to the membrane voltage and generally outwardly directed (de Campos Lima et al., 2019). Potassium conductances may alternatively be activated or modulated through various intracellular signal transduction pathways activated by ligand binding to various neurolemmal metabotropic neuromodulator receptors (Gerber and Gähwiler, 1994; Gerber et al., 1992; Raymond and Lapied, 1999; Saadi et al., 2012).
GABA ligand binding to GABABergic metabotropic receptors (Christie and North, 1988; Lacey et al., 1988; Li and Guyenet, 1996; Madden and Johnson, 1998; Wagner and Dekin, 1993) generates a slow activation of membrane potassium conductances (Best et al., 2007; Breton and Stuart, 2017; Christie and North, 1988; Gray and Green, 1987; Lacey et al., 1988; Li and Guyenet, 1996; Liu et al., 2012; Pfeiffer and Zhang, 2007; Saint et al., 1990; Schweitzer et al., 2004; Wagner and Dekin, 1993, 1997). There exist several types and classes of these GABABergic augmented potassium conductances, broadly designated according to barium and cesium sensitivity and specifically according to electrophysiological properties elucidated by intracellular, whole-cell patch clamp, and ion channel clamp recordings, in current-clamp or voltage-clamp mode (Breton and Stuart, 2017; Lacey et al., 1988; Li and Guyenet, 1996; Schweitzer et al., 2004; Wagner and Dekin, 1993). The GABABergic linked potassium conductances share in commonality activation by metabotropic G protein-mediated signaling (Callaway et al., 1992; Pfeiffer and Zhang, 2007).
The works of Wagner and Dekin (1993, 1997) have proven quite illuminative in thoroughly describing and characterizing potassium conductances within respiratory-related units, alternately characterizing the channel kinetics and electrophysiological properties of barium sensitive and barium insensitive potassium channels. These authors demonstrated the existence of a barium sensitive outward rectifying potassium current inhibited by the chemical mediator cAMP and sharing similar kinetics with S type potassium channels, present and mediating neuroplasticity in the mollusc Aplysia (Kandel, 1979; Wagner and Dekin, 1997). Wagner and Dekin (1993) also specifically characterized the electrophysiological properties and kinetics of a GABAB receptor-activated barium and cesium insensitive outward rectifier potassium channel termed GBac in respiratory-related premotoneurons. The channel was baclofen activated, exhibited outward rectification, and demonstrated a monochannel conductance of 100 picosiemens (pS). Reversal potential shift varying in accordance, and proportionally, with the concentration of extracellular potassium indicated the channel was predominantly selective for transmitting and conducting these ions, sharing similar biophysical properties to the cAMP inhibited S type potassium channel in Aplysia. The works of Wagner and Dekin (1993, 1997) thus contemporaneously characterized GABABergic modulated potassium channel molecular biophysics within respiratory-related units of the brainstem, evidencing conservation of the same biomolecular and ionic channel circuitry regulating membrane potential in neural respiratory circuitry and mediating neuroplasticity evolutionarily conserved from Aplysia, according to the pivotal works of Emeritus Professor Dr. Eric R. Kandel elucidating the neuronal cellular basis of learning. (N.B. Emeritus Professor Dr. Eric R. Kandel was awarded the Nobel Prize for discovering and elucidating short term facilitation and long term potentiation and depression in the mollusc Aplysia (Kandel, 1979; Kandel et al., 2000). These mechanisms collectively constitute the fundamental and essential biomolecular basis for neuronal learning, concurrent with the yet to be published discovery of the same by the works of Emeritus Professor Dr. Vitaliy Marchenko describing long term potentiation in the trigeminal pathway in mammals in Bogomoltez Institute in Kiev, Ukraine and various investigators in hippocampal formation in later years). The works of Pfeiffer and Zhang (2007) extended the findings of Wagner and Dekin (1993, 1997). These authors specifically characterized the electrophysiological and biophysical properties of a barium sensitive outward rectifying potassium conductance exhibiting a reversal potential of -78 mV in whole-cell patch clamp recordings of respiratory-related units from ventral respiratory column neurons in brainstem slices of mice. The GABAB antagonist CGP55845A inhibited the conductance. At each postnatal age, baclofen mediated GABABergic activation generated dose-dependent amplifications of the barium sensitive outward rectifier potassium conductance and these effects exhibited greater potency and efficacy contemporaneous with and paralleling increasing developmental maturation. For example, treatment with 1 μM baclofen in P2 (postnatal day 2) neonatal mouse brainstem slices generated hypoglossal bradypnea and a milder decrement of the hypoglossal burst amplitude, with rhythm abolition occurring in response to administration of 5 μM baclofen. In P9 neonatal rats, 1 μM baclofen proved sufficiently effective to generate respiratory rhythm abolition of hypoglossal neural discharge. Thus, baclofen exhibited age varying effects upon brainstem respiratory-related neuronal current density drug dose curves.
Thus, in general, ligand binding of GABA to the extracellular domain of GABAB receptors located within the postsynaptic terminal contemporaneously affects the metabotropic mediated augmentation of potassium ionic conductance (Breton and Stuart, 2017; Pfeiffer and Zhang, 2007; Wagner and Dekin, 1993) and attenuation of calcium ionic conductances (Cai et al., 2018; Mizuta et al., 2008; Li and Guyenet, 1995; Zhang et al., 1999). This is contrasted with the effects of GABA ligand binding to presynaptically located GABABergic receptors, which generally generates an attenuation of voltage-gated calcium ionic conductances, preventing calcium-mediated synaptovesicular fusion and neurotransmitter release into the synaptic cleft. The presynaptic GABABergic biomolecular machinery thus exerts a modulatory influence upon GABAB mediated hyperpolarization of the postsynaptic neuron. The GABABergic linked and modulable calcium currents of the neuron are generally thus broadly classified according to an activational threshold, designated alternately as low voltage-activated and high voltage-activated calcium channels (Chen and van den Pol, 1998; Harayama et al., 1998; Marchetti et al., 1991; Scholz and Miller, 1991). The high voltage-activated channels include P/Q type (Chen and van den Pol, 1998), N-type (Cai et al., 2018; Callaghan et al., 2008; Delaney and Crane, 2016; Huynh et al., 2015; Liu et al., 2018; Menon-Johansson, 1993; Sun and Chiu, 1999), and L type (Booker et al., 2018; Bray and Mynlieff, 2011; Maguire et al., 1989) channels, readily distinguished by biomolecular structure and electrophysiological properties. The L and N types of high voltage-activated calcium channels contribute to the oscillatory discharge of spontaneously bursting units involved in respiratory rhythm generation and pattern formation (Onimaru et al., 1996; Richter et al., 1993; Rybak et al., 1997), with the P and N types of high voltage-activated calcium channels contributing to modulation of neurosynaptic transmission (Dolphin et al., 1993; Wright and Angus, 1996). The low voltage-activated calcium channel mediates subthreshold membrane depolarizations, bringing the membrane voltage closer to action potential discharge threshold, and amplifies the excitability of neurons to incoming inputs.
Zhang and colleagues (Zhang et al., 1999) provide us with perhaps the most comprehensive, thorough, and detailed study evaluating the effects of GABABergic agonism upon the activities of the different modulated calcium currents constituting and contributing to the generation of neuronal transmission and electrosynaptic transduction in preBötzinger complex units. The different isoforms of these calcium channels (Chen and van den Pol, 1998; Harayama et al., 1998; Marchetti et al., 1991; Scholz and Miller, 1991) exhibit varying developmental kinetics and differential patterns of expressivity and activation in response to treatment with GABABergic agonists, providing a mechanistic basis through which GABABergic transmission differentially modulates electrophysiologically distinct elements of respiratory rhythm generation and pattern formation (Booker et al., 2018; Bray and Mynlieff, 2011; Cai et al., 2018; Callaghan et al., 2008; Chen and van den Pol, 1998; Delaney and Crane, 2016; Liu et al., 2018; Maguire et al., 1989; Menon-Johansson, 1993; Sun and Chiu, 1999). For example, in respiratory related brainstem zones, the high voltage-activated L type calcium channel exhibits developmental increases during the first two weeks of neonatal development, with the N-type channel becoming expressed during the second week of neonatal development. Though the ionic channel membrane density exhibits rapid and dynamic changes during development designed to adapt the requirements of inhibitory neurotransmission by the brainstem respiratory circuitry, the biophysical properties of these channels are highly conserved features and generally unalterable.
The GABAB agonist baclofen exerts dose-dependent effects upon the respiratory rhythm in mouse brainstem slices. GABABergic agonists generate activation of low voltage-activated calcium channels during early neonatal life, with the effect rapidly becoming inhibitory with increasing developmental maturation. There also exists a developmental dependent increase in the potency of GABABergic mediated inhibition of the low voltage-activated calcium channels varying according to age, contrasted with baclofen generated inhibition of the high voltage-activated calcium channels, which generally remains a constant feature in early and late neonatal development, with age invariant potency and efficacy. This general pattern is variably and differentially recapitulated elsewhere in the cerebrum, with developmental decreases of L type calcium conductances in hippocampal neurons (Thompson and Wong, 1991) and developmental increases of L type calcium conductances in thalamocortical neurons (Pirchio et al., 1990). Though the density of different types of calcium channels exhibits changes varying according to neonatal development, the biophysical and transduction properties of the channels proper demonstrate stability.
Thus, the collective results and findings of in vitro studies indicate powerful respiratory rhythm modulation by, though persistence in response to, bicuculline mediated GABAAergic and strychnine mediated glycinergic antagonism (Brockhaus and Ballanyi, 1998; Feldman and Smith, 1989; Murakoshi et al., 1985; Onimaru et al., 1990; Smith and Feldman, 1987). The works of Brockhaus and Ballanyi (1998) contemporaneously provided several powerful and beneficial insights into the contribution of both fast and slow neuromodulatory inhibitory synaptic transmission to respiratory rhythm generation and pattern formation. Respiratory rhythm persisted despite treatment with broad-spectrum pharmacological antagonists of GABAAergic and glycinergic signaling in the in vitro brainstem spinal cord preparation of the neonatal rat (Brockhaus and Ballanyi, 1998). Respiratory rhythmic discharge often persists in brainstem slice preparations containing the preBötzC despite lacking full network connectivity and in the presence of synaptic antagonism of fast inhibitory synaptic transmission (Holtman and King, 1988; Schmid et al., 1991; Smith and Feldman, 1987). These results would seem to suggest and support a pacemaker mechanism generating fictive neural breathing in these models. Alternatively, rhythmic discharge in preBötzC slices may be explained by basic neurobiological properties of the neurolemmal ion channels transducing persistent cationic currents, Nernst electrochemical potentials, and resting membrane voltage more closely approximated to action potential threshold (Anderson and Ramirez, 2017). High potassium concentrations utilized in these in vitro preparations may depolarize the membrane voltage towards action potential threshold sufficiently to generate spontaneous bursting activity manifest in recordings of neural respiratory discharge (Rybak et al., 2003a) by reducing the threshold of chemical disinhibition mediated acceleration of oscillator frequency, challenging the central interpretations of these studies.
Early studies evaluating this topic conducted in vivo continued to support a model of breathing in which pacemaker mechanisms may prove wholly sufficient to generate the breathing rhythm (Janczewski et al., 2013). Microinjections of bicuculline and strychnine in preBötzinger and Bötzinger complexes in the spontaneously breathing anesthetized preparation of the in vivo adult rat were shown to modify the respiratory rhythm and pattern evidenced in diaphragmatic electromyogram recordings in the vagus intact condition, though had no effects on breathing in vagotomized animals. These results were interpreted to indicate fast inhibitory neurotransductive modulation of the Hering Breuer reflex, though the ultimate neural respiratory rhythm independence of network mechanisms (Janczewski et al., 2013). Accordingly, investigators have continued to develop and espouse models whereby breathing may be independently generated wholly by the activity of spontaneous oscillators. These rhythm generators are distributed diffusely, though discretely, throughout the pons (Bonis et al., 2010), medulla (Anderson et al., 2016; Ramirez and Baertsch, 2019; Smith et al., 1991), and upper cervical spinal cord (Aoki et al., 1978, 1980; Ghali and Marchenko, 2016a) and include the preBötzC (Malheiros Lima et al., 2018; Morgado-Valle and Beltran-Parrazal, 2017; Smith et al., 1991), pFRG (Ikeda et al., 2019; Onimaru and Dutschmann, 2012; Onimaru et al., 1987, 1988), pontine respiratory group (Bonis et al., 2010), and upper cervical respiratory group (UCRG) (Aoki et al., 1978, 1980; Ghali and Marchenko, 2016a). A recently described spontaneous oscillator located rostrodorsomedial to the rostral pole of the nucleus ambiguus, termed the postinspiratory complex (Anderson and Ramirez, 2017; Anderson et al., 2016), may also contribute prominently to respiratory rhythm generation. Oscillator formation of respiratory rhythm is most elegantly and recently reviewed by Ramirez and Baertsch (2019) and Anderson and Ramirez (2017). We thus suggest the necessity of pacemaker mechanisms to respiratory rhythmogenesis in vivo may be specifically determined and interrogated by selectively or contemporaneously performing bilateral microinjections of the persistent sodium channel antagonist riluzole, the voltage-gated calcium channel antagonist amlodipine, and/or the calcium-activated nonselective cationic current antagonist cadmium in preBötzC, BötzC, RTN/pFRG, and/or PiCO.
The critical deficiency of breathing models predicated exclusively on pacemaker mechanisms to explain the generation of rhythmicity rests upon the necessity for inhibition to terminate the inspiratory epoch, generate expiratory segmentation, and thus resolve triphasic eupnea into inspiratory, postinspiratory, and late expiratory phases (Marchenko et al., 2016). Reciprocal mutual inhibition amongst Bötzinger complex dec post-I (Ezure and Manabe, 1988) and aug late-E (Jiang and Lipski, 1990) units underlies expiratory segmentation (Bianchi et al., 1995; Marchenko et al., 2016; Ghali and Marchenko, 2016b). Specifically, the sequential activity of medullary late inspiratory units (Cohen et al., 1993) and Bötzinger complex glycinergic dec post-I units (Ezure and Manabe, 1988) mediates the inspiratory off switching mechanism (Okazaki et al., 2001; Yamazaki et al., 2000). GABAergic and glycinergic antagonism powerfully modulates, though does not abolish, neural respiratory rhythmic activity in arterially perfused in situ (Dutschmann and Paton, 2002; Hayashi and Lipski, 1992; St John and Paton, 2002) and in vivo anesthetized (Hedner et al., 1981, 1984; Schmid et al., 1989, 1991a,b; Wessberg et al., 1983) preparations, typically generating tachypnea and/or hyperpnea. Thus, authors continued to identify the need to utilize in vivo preparations exhibiting full brainstem network connectivity utilizing localized microinjections in order to more precisely and specifically interrogate the role of fast inhibitory synaptic neurotransmission in generating different modes of observed neural respiratory patterns, a concept having permeated the ethos and zeitgeist of discussions in respiratory neurophysiology and advocated by Emeritus Professors Dr. V. Marchenko, J.C. Smith, I.A. Rybak, and Y.I. Molkov (Marchenko et al., 2016; Molkov et al., 2017; Rybak et al., 2014; Smith et al., 2009).
Modulation of GABAAergic, GABABergic, and glycinergic signaling in BötzC and preBötzC utilizing pressure microinjections of pharmacological agaonists and antagonists in anesthetized rabbits generates powerful effects upon respiratory rhythm, variably including tachypnea or bradypnea, hyperpnea or hypopnea, generation of biphasic patterns of discharge with alternating amplitude, and complete abolition of breathing (Bongianni et al., 2010). The influences of anesthesia may differentially suppress the discharge of sets of units within the preBötzC and the active population of units within preBötzC may differ substantially between anesthetized and unanesthetized decerebrate conditions (personal communication, Emeritus Professor Dr. Vitaliy Marchenko). Thus, Marchenko et al. (2016) demonstrated various powerful alterations or abolition of neural respiratory rhythmicity following microinjections of specific agonists and antagonists of GABAAergic and glycinergic signaling in BötzC and preBötzC in both anesthetized adult rats and unanesthetized decerebrate juvenile Sprague Dawley rats (Figs. 8-10). Specifically, combined local pressure microinjections of gabazine and strychnine in the preBötzC elicited increases in neural respiratory frequency, concurrent decreases of phrenic nerve amplitude, loss of post-I discharge in cervical vagus neurogram, and a reduction in expiratory duration. Combined gabazine and strychnine microinjections in the BötzC generated reductions of neural respiratory frequency and phrenic neurogram burst amplitude, loss of post-I discharge in cervical vagus neurogram, and increased expiratory duration, transitioning into apnea. Muscimol microinjections in the BötzC amplified neural respiratory frequency, which was readily reversed with gabazine microinjections. The sites of microinjection and drug concentrations were comparable to those used by Janczewski et al. (2013) in vagus intact and vagotomized anesthetized adult rats, indicating the findings of the present study are due to physiological effects rather than differences in microinjection protocol. Contemporaneous changes were observed in the arterial pressure in response to treatment with gabazine and/or strychnine, with pressor effects generated by microinjections in the BötzC likely attributable to effects upon the adjacent rostral ventrolateral medullary presympathetic bulbospinal units, and hypotension with microinjections in preBötzC, likely due to disinhibition of CVLM GABAergic units providing inhibitory modulation of RVLM presympathetic units. Antagonism of BötzC inhibitory neurotransmission generates apnea and BötzC muscimol silencing augments neural respiratory frequency, transitioning into tonic discharge (Bongianni et al., 2010; Marchenko et al., 2016). Accordingly, it is important to note the frequently utilized GABAAergic antagonist bicuculline also inhibits calcium activated potassium channels, blunting outwardly directed potassium current and thus generating nonspecific neuronal depolarization. Thus, gabazine represents a more specific and pure antagonist of GABAAergic signaling.
 Figure 8.
Figure 8.Extracellular recordings of preBötzinger and Bötzinger complex units prior to and following glutamate microinjections in vivo. A: Multiunit preBötzinger complex activity evidences biphasically augmenting preinspiratory discharge and smoothly augmenting inspiratory discharge relative to phrenic neurogram activity. The preBötzinger complex augmenting spatiotemporal burst profile of preinspiratory and inspiraotry activities exhibit differential kinetics with a more rapid rise of neuronal discharge frequency during the inspiratory compared with preinspiratory phase. B: Bötzinger complex multiunit activity demonstrates postinspiratory and augmenting late expiratory activity relative to the phrenic neurogram. C: Glutamate microinjections in the preBötzinger complex generate reductions in respiratory frequency accounted for predominantly by an increase in expiratory duration and an increase in phrenic nerve amplitude, as well as an initial hypotension and subsequent slight pressor effect, likely as a consequence of stimulation of GABAergic inhibitory propriobulbar interneurons within the rostral aspect of the caudal ventrolateral medulla. D: Glutamate microinjections in the Bötzinger complex generate apnea, likely mediated by activation of Bötzinger complex glycinergic decrementing postinspiratory neurons and GABAergic augmenting late expiratory neurons generating a complete inhibition of preBötzinger complex inspiratory units. Concurrent pressor effects likely result from stimulation of rostral ventrolateral medullary presympathetic units. Modified with permission from Fig. 4 of Marchenko et al. (2016).
 Figure 9.
Figure 9.Effects of gabazine and strychnine microinjections in preBötzinger complex on respiratory rhythm in vivo. Contemporaneous gabazine and strychnine microinjections in preBötzinger complex generates increases in phrenic nerve frequency and decrease in the phrenic nerve amplitude. B: Inspiratory duration decreases. C: Expiratory duration decreases. D: Phrenic nerve frequency increases. E: Phrenic nerve burst amplitude decreases. PN, phrenic nerve; TE, expiratory duration; TI, inspiratory duration; fR, frequency of respiration; integrated PN, integrated phrenic nerve amplitude. Modified with permission from Fig. 5 of Marchenko et al. (2016).
 Figure 10.
Figure 10.Effects of gabazine and strychnine microinjections in Bötzinger complex on respiratory rhythm in vivo. Contemporaneous gabazine and strychnine microinjections in Bötzinger complex generates decreases in phrenic nerve frequency and burst amplitude with a delayed transitory apnea. B: Inspiratory duration remains stable. C: Expiratory duration increases. D: Phrenic nerve frequency decreases. E: Phrenic nerve burst amplitude decreases. PN, phrenic nerve; TE, expiratory duration; TI, inspiratory duration; fR, frequency of respiration; integrated PN, integrated phrenic nerve amplitude. Modified with permission from Fig. 6 of Marchenko et al. (2016).
The preponderance of studies investigating GABAB receptor mediated modulation of the neural respiratory output have thus utilized in vitro neonatal mouse (Pfeiffer and Zhang, 2007; Zhang et al., 2002) or neonatal rat brainstem slice or brainstem spinal cord preparations (Brockhaus and Ballanyi, 1998; Johnson et al., 1996). A few studies have characterized the effects of GABABergic signaling upon the neural respiratory circuitry in vivo in rabbits (Schmid et al., 1989) and cats (Pierrefiche et al., 1993). Augmentation of GABABergic signaling utilizing pharmacological agonists characteristically effects hypopnea and bradypnea in neural respiratory discharges, an effect consistently demonstrated across several studies and preparation types in the in vitro neonatal mouse (Zhang et al., 2002; Pfeiffer and Zhang, 2007), in vitro neonatal rat (Johnson et al., 1996; Brockhaus and Ballanyi, 1998; Feldman and Smith, 1989), and in vivo unanesthetized decerebrate cat (Pierrefiche et al., 1993). In neonatal mouse brainstem slices, baclofen generates dose-dependent reductions of the respiratory rhythm and frequency and attenuates preBötzC inspiratory neuronal discharge frequency, with age variant effect modification. Baclofen mediated neural bradypnea was prevented by treatment with the GABAB receptor selective antagonist phaclofen (Feldman and Smith, 1989). In the in vitro brainstem spinal cord preparation of the neonatal rat, treatment with the GABABergic agonist baclofen was shown to generated respiratory-related neuronal membrane hyperpolarization, an effect mediated by amplification of a potassium conductance exhibiting a reversal potential of -88 mV (Brockhaus and Ballanyi, 1998). Baclofen mediated reductions of neural respiratory discharge frequency and membrane voltage hyperpolarization of the neuronal membrane were prevented by treatment with the potent competitive reversible GABABergic antagonist 2-hydroxysaclofen, without effects upon respiratory-related IPSPs.
The effect of an agonist to exert and generate a physiological effect in measured variabilities evidences the presence of functional receptors coupled to signal transduction mechanisms generating some measurable physiological effect. Thus, the use of pharmacological antagonists more precisely informs our understanding of native signaling in forming a physiological output. Accordingly, Zhang et al. (1999, 2002) provide data evaluating the contribution of GABABergic signaling to respiratory rhythm generation and pattern formation in vitro, through the precise and elegant use of agonists and antagonists of GABAA and GABAB receptors in order to precisely dissect the functional expressivity and native contribution of these signaling mechanisms in breathing generation. Zhang et al. (2002) demonstrated augmentations of respiratory related output and blunting of GABABergic agonist-mediated membrane voltage hyperpolarization and neural bradypnea in the in vitro preparation of neonatal mouse brainstem slices in response to treatment with the GABABergic antagonist CGP55845A. These results evidenced native and constitutive GABABergic modulation of the neural activity. Slopes of current density versus baclofen concentration curves progressively increased according to age, evidencing the developmental upregulation of receptor expression.
In the in vitro preparation of mouse brainstem slices, nipecotic acid-mediated antagonism of synaptic cleft reuptake of GABA generated neural bradypnea in both young and old neonatal mice (Zhang et al., 2002). These effects were reversed by treatment with CGP55845A in early neonates and either bicuculline or CGP55845A in older neonates. Specifically, GABABergic antagonism generated dose-dependent amplifications of neural respiratory frequency and amplitude in hypoglossal nerve discharge in P1 neonates and decreases of neural respiratory frequency in P9 slices (Fig. 11), evidencing native differential age variant effects of these treatments upon respiratory rhythm generation and pattern formation (Zhang et al., 2002). The age-dependent variation of the directionality and magnitude of effects elicited by GABABergic antagonism upon respiratory frequency was gradual.
 Figure 11.
Figure 11.GABAB receptor antagonism effects upon respiratory burst frequency across development. A and B: Age varying effects of CGP55845A (40 nM) on burst discharge frequency in brainstem slices in vitro derived from P1 (A) and P9 (B) neonatal mice. C: Bar graph quantifying concentration varying effects of the GABABergic antagonist CGP55845A in P0-P3 (open bars, n = 10) and P7-15 (filled bars, n = 14) neonatal slices. Threshold concentration to generate physiological effects was 20 nM in P0-P3 and 5 nM in P7-P15 neonates. D: Plot demonstrating age-dependent reversal of the physiological effect of CGP55845A (40 nM) from excitatory to inhibitory (P4-P6). Symbols indicate individual experiments performed on an individual slice obtained from a mouse at the indicated ages. Modified with permission from Fig. 1 of Zhang et al. (2002).
Zhang and colleagues (Zhang et al., 2002) also investigated the neuronal effects of GABABergic modulation. Baclofen reduced inhibitory postsynaptic current (IPSC) amplitude in early neonates and decreased both IPSP frequency and amplitude in older neonates. CGP55845A (40 nM) amplified preBötzC inspiratory neuronal discharge frequency and duration in P1 and P7 neonates and abolished the response to baclofen (0.75 μM) in P0 and P9 neonates (Fig. 12). CGP55845A mediated neural tachypnea and hyperpnea in younger neonates was not modified by GABAAergic or glycinergic antagonism. In contrast, CGP55845A generated neural bradypnea in older neonates, an effect which was reversed by bicuculline GABAAergic, and strychnine glycinergic, antagonism. Thus, the demonstration of GABABergic antagonist mediated modulation of neural respiratory frequency provides robust and powerful evidence validating a native contribution of GABABergic signaling in mediating inhibitory synaptic transmission during baseline conditions (Zhang et al., 2002). These findings collectively illumine and support a model of GABABergic mediated inhibition in younger neonates with a gradual shift to fast inhibitory synaptic transmission in older neonates, complementing neural maturational changes occurring with chloride based neurotransmission in respiratory column nuclei (Ghali and Beshay, 2019).
 Figure 12.
Figure 12.Developmental variation of GABABergic antagonism effects upon inspiratory neuronal membrane voltage and discharge properties. A and B: Current clamp mode sample recordings demonstrate the effect of CGP55845A on preBötzinger complex inspiratory neuronal membrane voltage and discharge properties in P1 (A) and P7 (B) neonatal mouse brainstem slices. Lower traces represent integrated activity of rhythmic discharge derived from hypoglossal neurogram. Recordings demonstrate membrane voltage depolarization and burst duration shortening elicited in response to microinjections of CGP55845A. Hyperpolarizing DC current (A3, B3) reversed the effects of GABABergic antagonism on neurolemmal membrane voltage and inspiratory burst duration. In the P7 neonatal mouse slice, hyperpolarizing DC current injections did not generate reversals of the reduction in the depolarizing synaptic drive potential (arrows in panel B4). Traces A1, A3 and B1, B3 superimposed in A4 and B4, respectively. C: Current-clamp mode recordings demonstrate baclofen mediated hyperpolarization of the membrane voltage and abolishes rhythmic inspiratory neuronal bursting in P0 (C1, C2) and P9 (C3, C4) neonatal mouse brainstem slices. D: Bar graph quantifying the effects of CGP55845A upon neurolemmal membrane voltage in inspiratory preBötzinger complex units from P0-P3 (open bars, n = 12) and P7-15 (filled bars, n = 12) neonatal mouse brainstem slices. E: bar graph summarizing the effects of baclofen upon membrane voltage in preBötzinger complex inspiratory neurons from P0-P3 (open bars, n = 10) and Fig. 5 of Zhang et al. (2002).
Combined GABAAergic and glycinergic antagonism with bicuculline and strychnine in neonatal mouse brainstem slices was shown to powerfully augment neural respiratory discharge frequency in hypoglossal neurogram in older neonates, though did not modify bursting frequency in younger neonates, recapitulating the age dependent neuromaturational features of chloride-conductance based neurotransmission (Zhang et al., 2002). Reciprocally, and corroborative of the described conjectures and proposed model properties, the bicuculline/strychnine cocktail generated profound amplifications of the hypoglossal bursting frequency in older neonates, evidencing functional maturation of chloride conductance based neurotransmission (Zhang et al., 2002). In contrast, administering the GABABergic antagonist CGP55845A with the bicuculline/strychnine cocktail generated profound amplification of the hypoglossal bursting frequency in younger neonates, specifically underscoring the critical and predominant importance of GABABergic signaling in mediating inhibitory synanptic neuromodulation during very early neonatal maturation (Zhang et al., 2002).GABABergic antagonism mediated neural bradypnea in the in vitro neonatal rat, demonstrated by Feldman and Smith (1989), and in older neonatal mice, demonstrated by Zhang et al. (2002), presents a dilemma alternatively resolved by a pharmacological, biochemical, or composite explanation (Zhang et al., 2002). The effects of CGP55845A to generate neural tachypnea in very early neonates expectedly evidences native and constitutive GABABergic mediated inhibitory modulation of respiratory output, though neural bradypnea paradoxically occurring in response to treatment with this compound in the in vitro brainstem spinal cord preparation of the neonatal rat (Feldman and Smith, 1989) and older neonatal mice (Zhang et al., 2002) would seem to evidence GABABergic signaling mediates excitatory signaling in these experimental preparations and are challenging to resolve according to common theoretical considerations. We will nevertheless strive in endeavoring to provide a coherent explanation subject to experimental validation according to our best understanding of the GABABergic biomolecular machinery. Given the heterogeneity of ligand binding zones of the GABAB receptor, CGP55845A could alternatively and contemporaneously mediate antagonist, partial agonist, and/or inverse agonist effects. Alternatively, differential GABAB receptor splice variant expressivity could contribute to the age-variant heterogeneity of effects. Lending credence to these conjectures, treatment with the GABABergic agonist baclofen generated significant decreases in IPSC amplitude, though it was without effect on IPSC frequency in early neonates. In contrast, both the frequency and amplitude of spontaneous and miniature IPSCs were reduced by treatment with baclofen in older animals. These effects were likely mediated by actions upon GABAB receptors in the presynaptic and postsynaptic membranes to generate increases of potassium conductances and/or reductions of calcium conductance (Misgeld et al., 1995).
Let us assume for the sake of argument, CGP55845A exerts no specific age-variant differential effects upon the GABABergic biomolecular machinery across development. Accordingly, let us suppose CGP55845A acts as a pure pharmacological antagonist in both young and old neonates. We are thus forced to develop interpretations and extrapolations of the conflicting data predicated upon subtleties of network interactions. In this regard, we suggest during the intermediate neonatal period (P9 neonatal mice in Zhang et al. (2002)), the biomolecular machinery modulating and conveying chloride based conductances is in the process of undergoing a transition from generating Nernst electrochemical potentials of the chloride ion which are predominantly electropositive, to those which are predominantly electronegative, with respect to the resting membrane voltage. This renders the opening of chloride ion channels to mediate depolarizing effects upon membrane voltage during the late fetal and early neonatal periods with a gradual shift to generating hyperpolarizing effects upon membrane voltage. Thus, during the early periods of neuromaturational development, the amplitude and frequency of neural respiratory rhythmicity is thus principally generated by the preBötzinger complex, parafacial respiratory group, and putatively the postinspiratory complex oscillators, though network mechanisms are ccontemporaneously evolving to couple activity amongst the oscillators. The generation of synchrony and power within neural respiratory circuitry and amongst spontaneous oscillators critically requires inhibitory network interactions (Marchenko and Rogers, 2009). Accordingly, blunting GABAB receptor mediated neuromodulation, representing the principal mechanism of synaptic inhibition during this transitional period of neonatal development, effectively reduces network synchrony and consequently contemporaneously compromises oscillator frequency and the generation of spectral power within neural respiratory bursts.
The complexity and heterogeneity of effects generated by the GABABergic biomolecular machinery modulating the neural respiratory output is evidenced by differential sensitivity of the respiratory rhythm, abolished by quite low doses of baclofen (1 μM), compared with that of membrane conductance mediated synaptic transmission, requiring much greater doses of baclofen to augment potassium (20 μM) and inhibit calcium (30 μM) currents (Brockhaus and Ballanyi, 1998; Johnson et al., 1996; Zhang et al., 1999, 2002). These differences may putatively arise consequent to the contemporaneous presence of GABAB receptors within synaptic and extrasynaptic zones (Ritter et al., 2004), generating amplification of potassium conductances (Wagner and Dekin, 1993, 1997), and attenuation of calcium conductances.
Variable and often disparate effects of GABABergic antagonists upon the respiratory rhythm in vitro indicated the necessity for studies utilizing preparations preserving neural respiratory circuitry intact in order to precisely resolve the native contribution of GABABergic signaling to breathing in neonates, juveniles, and adults. Accordingly, Pierrefiche et al. (1993) investigated the contribution of metabotropic GABAB receptor-mediated modulation of the discharge of individual neurons and whole phrenic nerve in anesthetized, unanesthetized decerebrate, and freely behaving cats. Iontophoretic application of baclofen generated decreases of medullary respiratory-related neuronal discharge frequency, effects antagonized by the selective GABABergic antagonists CGP55845A and saclofen (Pierrefiche et al., 1993). Treatment with either GABABergic antagonist effected reciprocal and marked augmentations of respiratory-related neuronal frequency in the majority of units (Pierrefiche et al., 1993). Intravenously administered baclofen generated amplifications of phrenic inspiratory duration, reductions of phrenic nerve amplitude, neural bradypnea, or complete apnea (Pierrefiche et al., 1993). The baclofen mediated effects upon the inspiratory duration were thus comparable to MK801 mediated N-methyl-D-aspartate antagonism generated apneusis (Pierrefiche et al., 1993).
In rabbits, intracerebroventricular administration of the GABAB receptor antagonist phaclofen augmented the phrenic nerve amplitude, reduced inspiratory duration, and augmented expiratory duration (Schmid et al., 1989). In contrast, treatment with baclofen reduced phrenic nerve amplitude, an effect prevented by pretreatment with the GABABergic antagonist phaclofen. In rats under the influence of Nembutal anesthesia pretreated with lithium hydroxybutyrate, administration of euphylline generated differential effects upon the neural respiratory output varying according to the initial blood pressure and proportionally with the euphylline induced decrement of arterial pressure magnitude (Lebedeva et al., 2010). The authors thus presented evidence indicating GABAB receptor activation modulates the respiratory response to adenosine receptor antagonism (Lebedeva et al., 2010).
These findings collectively evidence the functional expressivity of GABABergic receptors and biomolecular machinery in neural respiratory circuitry of the brainstem and spinal cord across development. The contribution of GABABergic signaling to respiratory rhythmogenesis and pattern formation will be most specifically and thoroughly resolved by determining the effects of microinjections of pharmacological agonists and antagonists in either or both the BötzC and preBötzC bilaterally upon respiratory rhythm and pattern utilizing in vivo preparations of unanesthetized decerebrate animals. A precedent for the tremendous benefit and utility of such models was demonstrated by the works of Marchenko and colleagues (Ghali, 2015, 2019a; Ghali and Marchenko, 2016a,b; Marchenko and Rogers, 2006a,b, 2007, 2009; Marchenko et al., 2012). Haji et al., (1990) investigated the effects of neuronal GABAergic augmentation in unanesthetized decerebrate cats utilizing in vivo intracellular recordings. Treatment with antagonists of GABA reuptake generated medullary respiratory-related neurolemmal membrane voltage hyperpolarization.
GABABergic signaling represents a critical mechanism contributing to the modulation of basal neural respiratory discharge and various breathing reflexes (Pfeiffer and Zhang, 2007; Zhang et al., 2002). We shall take for the case of argument and example, a study conducted by Canning et al. (2012), who demonstrated potent antitussive effects of the GABAB agonists lesogaberan and 3-aminopropylphosphinic acid in guinea pigs. The use of these drugs may obviate the sedative and respiratory suppressant effects ensuant from treatment with the GABAB agonist baclofen, more commonly employed in clinical therapeutics. Baclofen and 3 aminopropylphosphinic acid reduced cough induced by intrathoracic tracheal stimulation in cats and attenuated cough induced by inhaled capsaicin in unanesthetized guinea pigs (Bolser et al., 1993). Callaway and King (1992) investigated the antitussive effects mediated by α2 adrenergic and GABABergic agonists in a citric acid-induced cough model in guinea pigs. Citric acid-induced cough and tidal volume augmentation were attenuated by inhaled 5-allyl-2-amino-5,6,7,8-tetrahydro-4H-thiazolo[4,5-d]azepine dihydrochloride (B-HT 920) and xylazine. The physiological effects of the former were prevented by prior intraperitoneal treatment with the α2 adrenergic antagonists yohimbine, though not with the muscarinic cholinergic antagonist atropine. Inhalation of GABA or baclofen prevented citric acid-mediated augmentation of tidal volume, though demonstrated no antitussive effects. GABAB receptors within the brainstem neural respiratory circuitry or those located in the lungs may thus alternately or contemporaneously mediate the antitussive effects of baclofen and other congener agonists. In this regard, Chapman et al. (1993) demonstrated the functional expressivity of GABAB receptors in lung tissue, generating and effecting disparate effects, including inhibition of neuronally induced cholinergic and tachykinin mediated smooth muscle contraction, microvascular leakage, anaphylactic bronchospasm, and cough. Thus, further studies will prove necessary in order to more thoroughly resolve, and dissect the mechanisms contributing to, the modulatory effects of GABABergic signaling upon cough and other respiratory reflexes. These effects may alternately or contemporaneously be mediated by modulation of central brainstem and/or airway epithelial GABAB receptors.
Several investigators have underscored and highlighted the critical requirement of inhibitory elements in generating triphasic eupnea in adults and during the neonatal period (Ramirez et al., 1997; Richter et al., 1999). However, chloride reversal potentials are depolarized in very early neonates due to the high expression of sodium-potassium chloride cotransporters (NKCC1) mediating the import of chloride ions and low expression of potassium chloride cotransporter (KCC2) mediating the export of chloride ions (Fig. 13). Since the functional expression of GABAA and glycine receptors precedes the neuromaturational transition of chloride reversal potentials to hyperpolarizing values (Ritter and Zhang, 2000), signaling through these pathways exerts excitatory effects upon membrane voltage during the fetal and early neonatal periods. Importantly, there exists a well demonstrated developmental downregulation of sodium-potassium chloride cotransporters and upregulation of potassium chloride cotransporters in several brain regions including brainstem neural respiratory circuitry. This neuromaturationally varying pattern of cation chloride cotransporter expressivity causes the Nernst electrochemical potential of chloride to be relatively depolarized during the late fetal and early neonatal periods and hyperpolarized during the late neonatal, juvenile, and adult periods. These developmental neuromaturational changes thus gradually shift the chloride reversal potential to more hyperpolarizing values with age (Liu and Wong-Riley, 2012; Lu et al., 1999; Sedmak et al., 2016; Wong-Riley and Liu, 2005), rendering signaling via these pathways to exert inhibitory effects upon the membrane voltage in older neonates, juveniles, and adults.
 Figure 13.
Figure 13.Membrane ion transporters and channels modulating intracellular chloride ion concentrations in neural cells. Activity of the sodium potassium adenosine triphosphatase generates a net outwardly positive electrochemical potential difference. The sodium potassium chloride cotransporter (NKCC1) mediates the electroneutral symport of these ions in a 1 : 1 : 2 ([Na+] : [K+] : [2Cl-]) molar ratio, whereas the potassium chloride cotransporter (KCC2) mediates the electroneutral export of these ions in a 1 : 1 ([K+] : [Cl-]) molar ratio. The KCC2 cotransproter thus counteracts inwardly directed leak potassium conductances through outwardly directed active extrusion, contributing to hyperpolarization of the membrane voltage. The relative balance of NKCC1 mediated Cl-import and KCC2 mediated Cl-exports determines the Nernst electrochemical potential of this ion. Net extrusion of Cl- generating Nerst electorchemical potential differences more electronegative compared with the resting membrane voltage renders the opening of chloride ion channels to generate hyperpolarizing inwardly directed negative Cl- flux. Net Cl- import generating Nerst electorchemical potential differences more electropositive compared with the resting membrane voltage renders the opening of chloride ion channels to generate depolarizing outwardly directed negative Cl- flux. Voltage gated chloride ion channels, ligand gated chloride ion channels, and volume regulated anion channels contribute to the import or export of neural chloride, according to the electrochemical driving force. Furthermore, bulk chloride movement occurs via chloride bicarbonate (Cl-/HCO3-) anion exchangers. These biophysical properties and electrophysiological dynamics play a critical role in neonatal respiratory rhythmogenesis. Modified with permission from Fig. 2 of Wilson and Mongin (2019).
As alluded to previously, during the course of neonatal development, there exists a high expression of the sodium-potassium chloride cotransporter NKCC1 mediating the cellular import of chloride and low expression of the potassium chloride cotransporter KCC2 mediating the cellular export of chloride, demonstrated in several brain regions (Liu et al., 2012; Lu et al., 1999; Ritter and Zhang, 2000; Sedmak et al., 2016; Wong-Riley and Liu, 2005) and specifically in medullary zones organizing and generating the respiratory rhythm and pattern (Fig. 14) (Liu and Wong-Riley, 2012). This causes the Nernst equilibrium potential for chloride to be depolarizing during the late fetal period and early neonatal development, rendering GABA and glycine ligand binding to the GABAA and glycine receptors to mediate a net outwardly directed flux of chloride ions, thus causing the transmembrane voltage difference to depolarize, converse to the condition in the adult, where the opening of these channels mediates a net inwardly directed chloride current (Ghali and Beshay, 2019). During development, there exists a gradual decrease in the expression of the chloride importing NKCC1 transporter and an increase in the expression of the chloride exporting KCC2 transporter, causing the chloride reversal potentials to shift from depolarizing towards progressively increasing hyperpolarizing values. However, the generation of normal eupnea critically requires the intact functionality of inhibitory network elements (Molkov et al., 2017; Smith et al., 2007, 2009). Though preBötzC spontaneously bursting pre-I units diffusely distributed throughout various oscillators in the brainstem may generate a respiratory rhythm without the intact functionality of inhibitory network elements (Ramirez and Baertsch, 2019; Smith et al., 1991), this rhythm is more rudimentary, typically consisting exclusively of decrementing inspiratory bursting, without the post-I or late-E activity which characterizes the normal eupneic rhythm and pattern described by Richter and colleagues (Richter, 1982; Richter et al., 1986). In seeking to reconcile the immaturity of the circuitry organizing fast inhibitory synaptic neurotransmission during early development and the necessity of inhibition to generate triphasic eupnea, we suggest a plausible and logical deduction is that GABABergic signaling, exhibiting slower kinetics in comparison to GABAAergic and glycinergic signaling, may represent an intermediary transitional mechanism mediating the inhibition necessary for network generation of the normal breathing rhythm (Ghali and Beshay, 2019). These neurochemical maturational properties evidence and imply some other mechanism distinct from chloride-based neurotransmission mediates the described inhibition. We thus suggest GABABergic signaling may represent the most critical mechanism of inhibitory neurotransmission during late fetal and very early neonatal development, prior to full biomolecular maturation of the neurochemical circuitry generates gradual shifts of chloride reversal potentials from depolarizing to hyperpolarizing (Fig. 14) (Ghali and Beshay, 2019; Paton and Richter, 1995; Ritter and Zhang, 2000; Wilson et al., 2004).
 Figure 14.
Figure 14.Neonatal changes in the expression patterns of NKCC1 and KCC2 in several brainstem regions. NKCC1 expression exhibits neuromaturational decreases in specific brainstem zones comprising the neural respiratory network. There are prominent decreases of NKCC1 expression in brainstem respiratory oscillators, premotoneuronal regions, and motoneuronal network nodes. There are also prominent age-dependent decreases of NKCC1 expression in brainstem zones which are not classically designated as specifically contributing to respiratory rhythmogenesis and pattern formation, indicating these neuromaturational dynamics of chloride conductance biomolecular machinery constitute a general mechanism utilized throughout the neuraxis. The age-dependent reduction in NKCC1 expression reduces the cellular capacity to import chloride ions, rendering KCC2 principally determinative of extracellular and intracellular chloride ion concentrations and the Nernst electrochemical potential. The composite electrical and chemical driving forces acting upon chloride ions and the resting membrane voltage collectively determine whether the opening of membrane chloride ion channels generates a net outwardly versus inwardly directed flux of these ions. NKCC1, sodium potassium chloride cotransporter; PBC, preBötzinger complex; Amb, nucleus ambiguus; XII, hypoglossal motor nucleus; NTSvl, ventrolateral nucleus tractus solitarius; RTN/pFRG, retrotrapezoid nucleus parafacial respiratory group complex; DMNX, dorsal motor nucleus of the vagus; IO, inferior olive, CN, cuneate nucleus. Modified with permission from Fig. 6 of Liu and Wong-Riley (2012).
GABABergic signaling thus represents a logical mediator generating the inhibitory transmission necessary to organize normal triphasic eupnea during the neonatal period preceding the full neurochemical maturation of GABAAergic and glycinergic biomolecular machinery mediating chloride conductance-based fast inhibitory neurotransmission. We assert GABABergic signaling, remaining independent of neurolemmal chloride reversal potentials and mediating effects through the activation of potassium conductances and downregulation of calcium conductances, likely represents the earliest functionally mature mechanism mediating and organizing the inhibitory neurotransmission requisite to generate the normal breathing rhythm during early development (Gahwiler and Brown, 1985; Misgeld et al., 1995; Newberry and Nicoll, 1985; Nicoll et al., 1990; Thalmann, 1988).
Thus, we suggest GABABergic metabotropic neuromodulatory transmission generates the inhibition necessary to couple spontaneously bursting oscillators with network elements and organize respiratory rhythm generation and pattern formation during the very early neonatal period (Pfeiffer and Zhang, 2007; Zhang et al., 1999, 2002).This mechanism precedes neurochemical maturational shifts of chloride reversal potentials from depolarizing towards hyperpolarizing values (Liu et al., 2012; Lu et al., 1999; Sedmak et al., 2016; Wong-Riley and Liu, 2005), permitting GABAAergic and glycinergic signaling to mediate fast inhibition within the neural respiratory circuitry during later neonatal and juvenile development (Ghali and Beshay, 2019), approximating the character of the network during the adult condition (Marchenko et al., 2016). Specifically, the current model for neural respiratory network maturation we describe herein thus critically implicates an initial role of postsynaptic GABABergic mediated inhibition, followed by maturation of presynaptic GABABergic inhibitory mechanisms modulating brainstem neural respiratory oscillators and network elements, including RTN/pFRG biphasic pre-I units, preBötzC spontaneously bursting pacemaker pre-I cells, Bötzinger complex dec post-I and aug late-E, and VRG aug-I neurons (Pfeiffer and Zhang, 2007; Zhang et al., 1999, 2002). Neurochemical developmental shifts of chloride reversal potentials from excitatory to inhibitory permits full maturation of fast inhibitory synaptic neurotransmission signaling via GABAA and glycine receptors (Zhang et al., 2002). The molecular mechanisms underlying the developmental maturation of fast inhibitory synaptic transmission in neural respiratory circuitry is accordingly well characterized (Liu et al., 2012).
Experimental treatment of neonatal brainstem slice and brainstem spinal cord preparations with GABABergic agonists reduces neural respiratory frequency and amplitude, generating neuronal membrane voltage hyperpolarization and with GABABergic antagonists variably augments the neural respiratory frequency and/or amplitude (Brockhaus and Ballanyi, 1998; Johnson et al., 1996; Pfeiffer and Zhang, 2007; Pierrefiche et al., 1993; Zhang et al., 2000, 2002). Specifically, treatment with GABABergic antagonists augmentations neural respiratory frequency in vitro in neonatal mouse brainstem slices (Zhang et al., 2002) and respiratory related neuronal and phrenic nerve frequency in vivo in anesthetized, decerebrate, and freely moving cats (Pierrefiche et al., 1993). Conversely and paradoxocially, treatment with GABABergic antagonists effect neural bradypnea in neonatal rat in vitro preparations (Feldman and Smith, 1989) and brainstem slices derived from older neonatal mice (Zhang et al., 2002). The mechanisms generating these differential effects of GABABergic antagonism upon the neural respiratory output demonstrated in neonatal in vitro preparations remain to be more precisely elucidated and explained. These results and findings thus clearly evidence a critical role of endogenous, spontaneous, and native GABABergic signaling in modulating the respiratory rhythm and pattern across a spectrum of development and preparation types and molecular developmental changes contributing to differential effects varying according to age.
The biomolecular machinery mediating GABABergic signaling evidences several unique neuromaturational features (Fritschy et al., 1999; Malitschek et al., 1998) distinct from those underlying the development of GABAAergic and glycinergic signaling (Liu et al., 2012; Lu et al., 1999; Paton and Richter, 1995; Ritter and Zhang, 2000; Sedmak et al., 2016; Wilson et al., 2004; Wong-Riley and Liu, 2005). Both neuronal transduction mechanisms exhibit progressive developmental amplifications of biomolecular sensitivity. Age variant effects of GABAAergic and glycinergic signaling are principally mediated through gradual developmental shifts of chloride reversal potentials, upregulation of postsynaptic receptors, and differential expression of receptor subunits (Liu and Wong-Riley, 2012). Maturational changes linked to GABABergic synaptic neuromodulation involve differential receptor expression (Malitschek et al., 1998), upregulation of calcium ionic channel conductance, improved fidelity of receptor ion channel coupling (Elsen and Ramirez, 1998; Misgeld et al., 1995; Zhang et al., 1999), and variant differential expression of biomolecular splice variants (Fritschy et al., 1999), during different periods of development.
Neonates characteristically express the GABAB1a splice variant and adults express the GABAB1b splice variant (Fritschy et al., 1999). This mechanism putatively contributes to dynamically mediating the developmental changes of receptor-ligand binding affinity and effector coupling. The fidelity of receptor effector coupling contributes importantly to the effects of GABABergic receptor activation upon membrane ionic conductances. For example, calcium channels present in the neurolemmal membrane not effectively coupled to the GABAB receptor signal transduction mechanisms may not effectively open in response to ligand binding (Elsen and Ramirez, 1998; Misgeld et al., 1995; Zhang et al., 1999). Furthermore, the expression patterns of GABABergic modulated membrane ion channels exhibit rapid and dynamic changes during the initial few weeks of neonatal development (Zhang et al., 1999). Membrane density of high voltage-activated L type calcium channels gradually increases during the first two weeks of neonatal development, with cell surface expression of N type calcium channels increasing starting during the second week of development (Zhang et al., 1999). GABABergic agonist potency and efficacy upon the high voltage-activated calcium ion channels remain relatively stable (Zhang et al., 1999). In contrast, GABABergic agonists mediate inhibition of low voltage-activated calcium ion channels during early development, with the effect rapidly becoming stimulatory, dose-dependent, and age variant within a few days of birth (Zhang et al., 1999).
The collective efforts of several studies have thus clearly demonstrated a critical role of GABABergic signaling in modulating brainstem neural respiratory, sympathetic, and parasympathetic circuitry (Lemus et al., 2008; Li and Guyenet, 1995; Wang et al., 2010). Wang et al. (2010) investigated and elucidated the relative contributions of GABAA and GABAB receptor activation upon the inhibitory dynamics of vagal afferent neurons in anesthetized rats (Wang et al., 2010). Treatment with the GABAAergic antagonist bicuculline delayed the onset and prolonged the duration of inhibition of vagosensitive neurons within NTS (Wang et al., 2010). Treatment with the GABABergic antagonist CGP35348 augmented the duration of NTS unitary inhibition, though exhibited no effects upon the onset of inhibition (Wang et al., 2010). Treatment with GABAB antagonists generated no effects upon neuronal inhibitory dynamics (Wang et al., 2010). Intracellular recordings mechanistically evidenced a biphasic contribution to the observed inhibition with early and late reversal potentials correspondent to the Nernst electrochemical potentials of chloride (GABAA receptor channel pore) and potassium (GABAB receptor coupled metabotropically activated channel) (Wang et al., 2010).
Li et al. (1995) demonstrated inhibition of rostral ventrolateral medullary units (84 of 87) in the in vivo and in vitro slice preparations of the rat occurring in response to treatment with the GABABergic agonist baclofen, evidencing hyperpolarization in 13 of 17 units, decreases of input resistance in 12 of 16 units, and reduced spontaneous synaptic activity in 7 of 14 units. The effects were dose-dependent and effectively inhibited by the GABAB antagonists CGP45626A, CGP55845A, and 2-hydroxysaclofen. Iontophoretic application of baclofen generated prominent inhibition of barosensitive bulsospinal (15 of 16) and respiratory-related (7 of 7) units. Lemus et al. (2008) demonstrated a specific effect of GABABergic signaling in modulating glucose responses coordinated by NTS units. Treatment with agonists or antagonists of GABABergic, but not of GABAAergic, signaling specifically modulated the arterial blood glucose concentration and carotid body chemoreceptor stimulation mediated augmentation of the glucose concentration.
Thus, GABABergic signaling contemporaneously modulates IPSPs in RVLM and NTS units and NTS and carotid body chemoreceptor mediated effects on arterial blood glucose concentrations. In this regard, glucosensitive nucleus tractus solitarius units receiving afferent inputs from carotid body chemoreceptors are thus specifically under tonic inhibitory influence by GABABergic mechanisms. The rostral ventrolateral medullary presympathetic units providing the bulbospinal drive to sympathetic preganglionic neurons interestingly exhibit glucosensitive membrane voltage properties. These findings collectively indicate the existence of a complex network mechanism regulating glucose-mediated changes in sympathetic and parasympathetic outflows, powerfully influenced by fast inhibitory GABAAergic synaptic neurotransmission and slow inhibitory GABABergic synaptic neuromodulation. Further studies upon this topic will likely prove to be a fruitful endeavor revelatory of novel mechanisms and interactions amongst and between brainstem sympathetic oscillators and propriobulbar inhibitory circuitry.
In evaluating our data and a plethora of studies, we have concluded, supported by empirical and theoretical validation discoursed upon in the following discussion, that all motor networks comprised of motoneuronal and interneuronal conglomerates distributed within brainstem and spinal cord exhibit as an intrinsic property an inherent capacity to generate rhythmic and patterned motor activity (Ghali and Marchenko, 2016a). These motor networks may be computationally conceptualized as sequential groups of motoneurons coupled with excitatory and inhibitory propriobulbar and propriospinal interneurons rostrocaudally, locally, and cross segmentally (Molkov et al., 2017). These motor networks or zones receive descending bulbospinal and propriospinal, ascending propriospinal, and local phasic and tonic excitatory and inhibitory inputs and exhibit differential thresholds for the genesis of rhythmic activity. Motor networks are driven and synchronized by the lowest threshold spontaneously discharging oscillator projecting to and commonly coupling the motoneuronal pools. The pattern evidenced in the motor outputs reflects the integrity of inhibitory neurosynaptic transduction within the common source highest order oscillator (Marchenko et al., 2016). Rostrocaudal network reduction contemporaneously reduces inhibitory and excitatory network elements, though spares preBötzC spontaneously bursting units, preferentially favoring pacemaker mechanisms compared with network mechanisms (Fig. 15). A fully intact central oscillator network robustly supplied by oxygen generates triphasic eupnea. Supraspinal network oscillators compromised by strategically placed mechanical transection between specific network components, antagonism of fast inhibitory synaptic transmission, or severe hypoxia generate bell-shaped biphasic patterned (inspiratory and expiratory) or decrementing monophasic (inspiratory) rhythmic activity. In broad terms, network mechanisms mediating respiratory rhythmogenesis require the functional integrity of GABAergic and glycinergic inhibitory propriobulbar interneurons. In brief, preBötzC pre-I units inhibit and are, in turn, inhibited by BötzC dec post-I and aug late-E cells. The GABAAergic and glycinergic mechanisms are principal and the GABABergic mechanisms neuromodulatory. These cells require an abundant supply of oxygen and glucose in order to support the high metabolic energy demands and oxidative phosphorylation necessary to generate high-frequency synaptic transmission (spinal gasping is a perfect example of these concepts; Ghali and Marchenko, (2016a)). Reductions of tissue oxygen supply by ischemia, asphyxia, or anoxia compromises inhibitory network elements and thus preferentially favors pacemaker mechanisms to more prominently contribute to mediating rhythmic and patterned motor output (Ghali and Marchenko, 2016a; St John, 1990). Under conditions of energetic stress or oxygen deprivation, patterned output generally degenerates into biphasic and monophasic rhythmic activity. Thus, network mechanisms are operant during normoxic and hyperoxic conditions providing sufficient oxygen to support inhibitory network elements, generating robust triphasic eupnea. Pacemaker mechanisms become operant with severe oxygen deprivation, generating more rudimentary monophasic decrementing bursting activity (Smith et al., 2007).
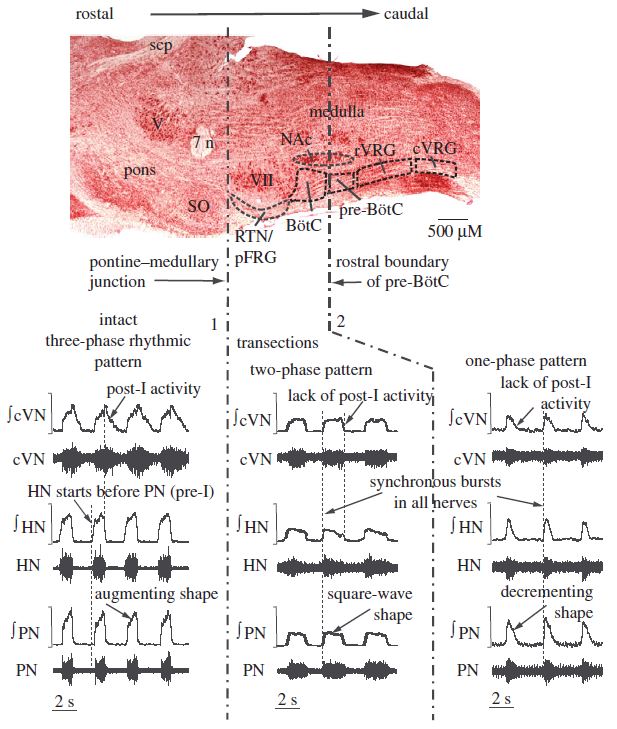 Figure 15.
Figure 15.Mammalian brainstem neural respiratory circuitry. Top panel demonstrates parasagittal slice through the ventrolateral pontomedullary respiratory column nuclei. The generation of the respiratory rhythm critically requires mutual and reciprocal inhibitory interactions between units in the Bötzinger and preBötzinger complexes. Normal triphasic eupnea consists of neural inspiration, postinspiration, and late expiration evident in the discharge patterns of central neurons and activity of peripheral respiratory related neurograms. Phrenic nerve discharge contains inspiratory and occasionally postinspiratory discharge. The hypoglossal nerve contains inspiratory discharge with variable amplitude under resting conditions and in unanesthetized conditions demonstrates preinspiratory activity, which becomes significantly more prominent with vagotomy and hypercapnia. The cervical vagus nerve evidences inspiratory and postinspiratory activity. Pontomedullary transection removing the tonic excitatory inputs of Kölliker-Fuse and medial parabrachial nucleus propriobulbar interneurons to Bötzinger complex glycinergic decrementing postinspiratory neurons, as well as tonic excitatory inputs provided by retrotrapezoid nucleus glutamatergic units to Bötzinger complex GABAergic augmenting late expiratory neurons, generates transitions from normal triphasic eupnea to a two phase breathing pattern consisting of inspiration and nonsegmented expiration. Transection between the Bötzinger and preBötzinger complex generates monophasic inspiratory bursting, lacking neural post-inspiration or expiration, bearing striking resemblance to gasping patterns in bulbospinal and cervically transected conditions. Thus, the progressive reduction of the neural respiratory circuitry sequentially removing inhibitory network elements gradually shifts the complexity of the classic triphasic eupneic rhythm. SO, superior olive; V, trigeminal nucleus; 7n, facial nucleus; RTN, retrotrapezoid nucleus; pFRG, parafacial respiratory group; scp, superior cerebellar peduncle; BötC, Bötzinger complex; pre-BötC, preBötzinger complex; rVRG, rostral ventral respiratory group; cVRG, caudal ventral respiratory group. Modified with permission from Fig. 1 of Smith et al. (2009).
A maximal electrical impulse delivered to the motor networks generates rhythmic or patterned bursting activity. In intact spinal preparations under normoxic and hyperoxic conditions, the pontomedullary respiratory network delivers rhythmically segmented high-frequency oscillatory drive to the brainstem and spinal respiratory-related motoneuronal pools (Marchenko et al., 2012). In cervicomedullary transected preparations in severely hypoxic conditions, anoxic depolarization drives the rhythmic bursting activity of a common oscillator in the upper cervical respiratory group or contemporaneously and synchronously drives the rhythmic bursting of spinal motor networks individually, in turn, synchronized by propriospinal excitatory interactions (Ghali and Marchenko, 2016a). In cervicomedullary transected preparations in normoxic and hyperoxic conditions, epidural spinal cord stimulation within the range of normal pattern of fast synchronous oscillations generates rhythmic bursting activity in phrenic nerve discharge supporting life-sustaining breathing (Bezdudnaya et al., 2018). We will provide and discuss several empirical findings further developing the model presupposed by and predicated upon these conjectures.
 Figure 16.
Figure 16.Decrementing pattern of asphyxia-induced bursting in phrenic nerve discharge in the high cervicomedullary (C1) transected unanesthetized decerebrate adult rat. A: Administration of 10% hypercapnia tests fail to elicit eupneic or any other activity in phrenic nerve discharge, neurophysiologically confirming complete bulbospinal transection of the brainstem respiratory central pattern generator from the phrenic motor circuitry. Asphyxic challenge performed immediately thereafter reveals the appearance of phasic activity in the phrenic neurogram superimposed on an increase in tonic discharge evidencing the existence of a spinal gasping generator. B-D: magnified views of insets in ‘A,’ ‘B,’ and ‘C,’ respectively. Asphyxia-induced bursts exhibit decrementing spatiotemporal dynamics highly reminiscent of classic brainstem gasping (lower right subpanel D). PhN, phrenic nerve; TP, tracheal pressure; CO2, end-tidal carbon dioxide. Modified with permission from Fig. 6 of Ghali and Marchenko (2016a).
We use as illuminative of our preceding conjectures the following empirical data in support of the same. Gasping consists of monophasic decrementing inspiratory rhythmic bursting with maximal discharge at onset definitionally lacking neural expiratory activity (Ghali and Marchenko, 2016a; Ramirez and Lieske, 2003; Richter, 2003; St John, 1990, 1996). The gasping mechanism was shown to utilize persistent sodium currents by Paton et al. (2006). Bilateral electrolytic lesions of the preBötzC or unilateral kainic acid lesions of the medullary lateral tegmental field effectively eliminate the capacity to generate gasping in response to severe oxygen deprivation (Fung et al., 1994; St John et al., 1984). Authors have thus classically designated the gasping center to reside within the preBötzC or medullary LTF, a zone also believed to originate sympathetic activity by several authors (Barman et al., 2019; Dampney, 2004; Ghali, 2017a; Marchenko and Sapru, 2003). However, we have previously demonstrated the emergence of monophasic decrementing rhythmic bursting in phrenic motor output in response to asphyxia in high cervically transected unanesthetized decerebrate rats (Ghali and Marchenko, 2016a), bearing a striking resemblance to the gasping rhythm evidenced in respiratory related nerve efferent neurograms in bulbospinal intact preparations (Marchenko and Rogers, 2006a). Why gasping should be completely abolished by very localized lesions to specific zones within the medulla, but not by complete bulbospinal dissociation via cervicomedullary transection presents quite the dilemma. This set of findings at apparent interexperimental contradistinction not only suggests the existence of spinal oscillators capable of generating true gasping or gasping like activity, but rather directs and guides us towards developing a finer and more detailed explanation of the mechanisms generating rhythmic motor output. The reconciliation of these empirical differences is principally illuminative and supportive of our preceding conjectures.
We suggest the preBötzC represents the oscillator exhibiting the lowest spontaneous activational threshold in response to severe oxygen deprivation and external perturbations. PreBötzC spontaneous bursting units discharge rhythmically via spontaneously depolarizing persistent sodium and calcium-activated nonselective cationic currents (Morgado-Valle et al., 2010; Rekling and Feldman, 1998; Smith et al., 1991; Toporikova and Butera, 2011; Winter et al., 2009). Membrane voltage approximation of threshold causes preBötzC cells to discharge powerfully and in unison. Anoxic recruitment of preBötzC units thus generates gasping (Ghali and Marchenko, 2016a; St John, 1990) and synchronizes efferent respiratory motor outputs throughout the rhombomyelic neuraxis. We assert, mechanistically, severe oxygen depletion causes preBötzC cells to discharge consequently to anoxic depolarization. Severe oxygen depletion reduces a cell’s capacity to effectively generate electron rich reducing equivalents (NADH, FADH2) necessary to generate high energy (adenosine triphosphate) phosphate bonds through oxidation-reduction reactions at multiunit complex proteins in the mitochondrial inner membrane. Adenosine triphosphate depletion compromises the functionality of neurolemmal sodium-potassium adenosine triphosphatase membrane pumps, reducing the electrogenic efflux of net positive current, and thus mediates a gradual depolarization of the membrane voltage, reducing the transmembrane potential and separation of charge, increasing the probability of spontaneous neuronal bursting. This mechanism contemporaneously causes metabolic exhaustion of inhibitory interneuronal elements and anoxically initiates the spontaneous bursting of preBötzC units mediated by low threshold persistent sodium and calcium-activated nonselective cationic currents. According to the presented model, severe hypoxia-induced bursting is initiated by anoxic depolarization, terminated by depletion of high energy phosphate bond reserves (e.g., creatine phosphate, adenosine triphosphate), and reinitiated by anaerobic glycolysis mediated restoration of the adenosine triphosphate pool. In this regard, neuronal glucose pools represent the critical limiting factor determining the duration the gasping rhythm may be effectively sustained by biomolecular mechanisms. A maximal impulse is delivered, the motor network discharges maximally, becomes depleted of adenosine triphosphate, and the burst terminates. Anaerobic glycolysis mediated restitution of adenosine triphosphate stores primes the motoneuronal pool to reinitiate bursting activity.
We assert, in the bulbospinal intact condition, the hyperpneic phase of the response to hypoxia represents acceleration of the network central pattern generator, primary apnea represents metabolic compromise of the inhibitory elements necessary to generate triphasic eupnea, bell-shaped transition bursts represent the network mechanism transiently discharging with a paucity of inhibitory elements, and decrementing monophasic bursts represent true gasping generated by anoxic depolarization of the lowest threshold highest order commonly synchronizing brainstem oscillator (e.g., preBötzC, medullary lateral tegmental field). The preBötzC represents the lowest threshold group of propriobulbar interneurons exhibiting extensive interconnectivity and influence upon respiratory motor outputs. Thus, according to our schema, the preBötzC and adjacent zones within the medullary LTF conceptually represent low threshold drivers and integrators of the gasping mechanism. In the bulbospinal intact condition with preserved descending phasic excitation and tonic inhibition, preBötzC spontaneously bursting units are powerfully driven by severe oxygen deprivation. This occurs directly via anoxic depolarization of the preBötzC units proper and via the loss of tonic inhibitory regulation by fast inhibitory synaptic neurotransmission. The preBötzC units, in turn, drive and monophasically synchronize brainstem and spinal cord motor outputs. In the cervicomedullary transected condition, spinal motor networks are alternately driven by the lowest threshold oscillator, presumptively localizing to the upper cervical spinal cord, or by the direct anoxic depolarization of each of the motoneuronal pools individually cross-segmentally synchronized by propriospinal relays. Well designed studies which may empirically validate the presented model would involve performing contemporaneous electrophysiological recordings and time frequency representation coherence analyses of multiple respiratory-related brainstems and spinal motoneuronal and neurogram outputs in bulbospinal intact and cervicomedullary transected conditions during normoxia, and in response to severe hypoxia, in order to more precisely distinguish among and preferentially validate these alternative hypotheses.
During neonatal development, there is high expression of cation chloride cotransporters mediating the cellular import of chloride (i.e., NKCC1) and low expression of cation chloride cotransporters mediating the cellular export of chloride (i.e., KCC2) (Dzhala et al. 2005; Liu et al., 2012; Lu et al., 1999; Ritter and Zhang, 2000; Sedmak et al., 2016; Wong-Riley and Liu, 2005). This causes the Nernst equilibrium potential for chloride to be hyperpolarizing during the late fetal period and early neonatal development, causing GABA and glycine ligand binding to the GABAA and glycine receptors, respectively, to generate a net outwardly directed flux of chloride anions, causing the membrane voltage to depolarize, drastically contrasted with the electrophysiological behavior of this signaling mechanismin the adult, where the opening of these channels mediates a net inwardly directed chloride current (Ghali and Beshay, 2019). With development, there is a gradual decrease in the expression of the chloride importing NKCC1 transporter and an increase in the expression of the chloride exporting KCC2 transporter, causing the chloride reversal potentials to gradually shift towards hyperpolarizing values. However, normal eupnea critically requires the intact functionality of brainstem inhibitory network elements in order to shape rVRG aug-I activity and generate post-I and late-E expiratory segmentation (Marchenko et al., 2016) and spinal local inhibitory network elements to modify the phasic and tonic components of the motoneuronal outputs (Marchenko et al., 2015). Though preBötzC and spontaneously bursting pre-I units diffusely distributed throughout various oscillators in the brainstem (Anderson and Ramirez, 2017; Anderson et al., 2016; Bonis et al., 2010; Ramirez and Baertsch, 2019; Smith et al., 1991, 2007, 2009) may generate respiratory rhythmicity independent of the intact functionality of inhibitory network components, these rhythms are significantly more rudimentary, typically consisting of exclusive inspiration (Smith et al., 1991), lacking the post-I or late-E phases which characterizes the normal eupneic rhythm and pattern described by Richter and colleagues (Richter, 1982; Richter et al., 1986). These empirically derived interpretations thus present quite the conundrum, or one may venture to surmise, scientific debacle. In seeking to reconcile these differences, specifically, the relative immaturity of the fast inhibitory synaptic neurotransmission circuitry in early development and the necessity of the same for the genesis of eupnea (Liu and Wong-Riley, 2012), a plausible and logical deduction suggests GABABergic signaling, slower in comparison to GABAAergic and glycinergic signaling, may represent an intermediary mechanism mediating the inhibition necessary for network generation of normal breathing. We may thus logically deduce the preponderance of evidence supports GABABergic signaling figures prominently in contributing to neonatal and adult respiratory rhythm generation and pattern formation (Ghali and Beshay, 2019).
We have thus made evaluated studies investigating the role of GABAergic signaling in respiratory rhythmogenesis and pattern formation (Pfeiffer and Zhang, 2007; Pierrefiche et al., 1993; Schmid et al., 1989; Zhang et al., 1999, 2002). The respiratory central pattern generator thus exhibits oscillator redundancy, utilizes inhibitory mechanisms, may reconfigure in response to various stressors, and can operate in different modes with physical or neurosynaptic network reduction. The central tenets and goals of our discourse are principally to suggest GABAAergic and glycinergic signaling are critically implicated in network models of respiratory rhythmogenesis and pattern formation. GABABergic signaling neuromodulates breathing output according to the slow kinetics of membrane channel effector mechanisms, though may represent the most critical mechanism of inhibitory neurotransmission during the very early neonatal development, prior to complete maturation of the neurochemical circuitry mediating membrane chloride conductances via a gradual shift of chloride reversal potentials from depolarizing to hyperpolarizing (Ghali and Beshay, 2019). Specifically, GABABergic activation amplifies postsynaptic membrane barium sensitive and insensitive potassium conductances (Wagner and Dekin, 1993, 1997) and attenuates presynaptic and postsynaptic calcium ionic conductances. Chloride reversal potential independence of GABABergic neurotransmission (Pfeiffer and Zhang, 2007; Wagner and Dekin, 1993, 1997) suggests this neurotransductive mechanism critically contributes to inhibitory neuromodulation in neonatal neural respiratory circuitry before full maturation of cotransporters mediating the cellular transport of chloride ions (Ghali and Beshay, 2019). Further studies will illumine our understanding of the role of GABAAergic and glycinergic synaptic neurotransmission and GABABergic synaptic neuromodulation in respiratory rhythmogenesis and pattern formation. Thoroughly resolving the network mechanisms generating rhythmic and patterned respiratory motor output informs our understanding of otherwise unexplainable emergent properties of physiological behavior of neural ensembles in cerebrum, brainstem, cerebellum, and spinal cord. Accordingly, in concluding his seminal work Principles of Neural Science, Emeritus Professor Dr. Eric R. Kandel provides a most wonderful description of consciousness reflecting an emergent property of complex neural network behavior amongst disparate cortical and thalamic circuitry (Kandel et al., 2000; Schwartz, 2000). The integrated and collective efforts of neurophysiologists and neurobiologists investigating and dissecting neural network mechanisms shall thus have as its Golden Grail and principal fruit the precise and elegant elucidation of mechanisms generating motor behavior, learning, and consciousness as complex emergent properties of neural network synchrony, dynamics, and synaptic plasticity.
Dedicated to my beloved father Dr. George Zaki Ghali, Emeritus Professor of Toxicology, and beloved mentor Dr. Vitaliy Marchenko, Emeritus Professor of Neurophysiology.
The author declares no conflict of interest.