


1 Brain Repair Group, Neuroscience department, School of Biosciences, Cardiff University, CF10 3AX Cardiff, UK
Academic Editor: Rafael Franco
Abstract
Advanced therapeutic medicinal products (ATMPs), including cell and gene therapies, are in development for Parkinson’s disease (PD). In many cases, the goal is to replace the lost dopamine (DA), which is anticipated to improve motor dysfunctions associated with DA loss. However, it is less clear the extent to which these therapeutic interventions may impact on the wide range of cognitive symptoms that manifest as the disease progresses. Although the accepted perception is that cognitive symptoms are predominately non-DAergic in origin, in this commentary, it is argued that several, specific cognitive processes, such as habit formation, working memory and reward processing, have been reported to be DA-dependent. Furthermore, there is evidence of DAergic medications modulating these behaviours in PD patients. Finally, the potential for cell and gene ATMPs to influence these symptoms is considered. It is concluded that DA replacement through ATMPs is likely to improve certain DA-dependent symptoms, but only sparse clinical data are currently available and the ability to precisely titrate DA transmission is likely to be complex.
Keywords
- Parkinson's disease
- Non-motor symptoms
- Dopamine
- Cell therapy
- Transplant
Parkinson’s disease (PD) affects over 6 million people in the world and prevalence is known to be rising [1]. The pathogenesis of PD is complex and multiple cellular processes have been implicated, including mitochondrial and lysosomal dysfunction, which are associated with the pathological aggregation of alpha-synuclein and the degeneration of midbrain dopamine (DA) neurons. Although therapeutic interventions are in development which aim to target these cellular processes, the majority of current treatments aim to replace the lost DA or DA signalling. The primary pharmacological treatments available in the clinic include Levodopa, a DA pro-drug, and DA agonists.
A number of advanced therapeutic medicinal products (ATMPs) are also in development and in active clinical trials, such as cell and gene therapies. Cell therapies involve the transplantation of live cells into the brain, using of preparations rich in DA progenitors, with the aim of replacing the lost DA neurons and restoring the nigrostriatal DAergic synapse. The proof-of-concept in this field was established using fetal DA cells, but current trials typically use human embryonic or induced pluripotent stem cells, which have been differentiated towards a DAergic fate (for comprehensive reviews, see [2, 3, 4, 5, 6]). Some gene therapy approaches involve the infusion of viral vectors into the caudate-putamen that express genes necessary for the biosynthesis of DA. Specifically, vectors harbouring L-DOPA synthesizing enzymes, tyrosine hydroxylase and guanosine-tri-phosphate-cyclohydrolase-1 (in combination with endogenous or vector-mediated aromatic L-amino acid decarboxylase) have been explored in preclinical and clinical settings [5, 7, 8, 9, 10]. Like the pharmacological interventions, these novel ATMPs also aim to replace the lost DA, but they have the advantage of (1) allowing targeted expression of DA in dennervated regions to avoid off-target effects and (2) inducing more constant, physiological release of DA, which is hypothesised to reduce the likelihood of developing motor side effects such as dyskinesias. Cell and gene ATMPs are well-established in their ability to improve motor dysfunctions in preclinical models of PD [11, 12] and many are currently in clinical trials [3, 5]. Thus, in this Commentary, the goal is to consider whether these ATMPs have the potential to impact a broader range of symptoms than just motor impairments, with a specific focus on the DA-dependent cognitive impairments in PD.
Although the primary hallmarks of PD are classic motor symptoms, there has recently been greater recognition of the wide variety of non-motor symptoms that manifest across the course of the disease and the significant impact of these non-motor symptoms on quality of life [13, 14]. Non-motor symptoms incorporate a wide variety of dysfunctions, ranging from cognitive and neuropsychiatric impairments to gastrointestinal dysregulation and sleep disturbances [15, 16, 17, 18, 19, 20]. Systematic tracking of symptom onset has demonstrated that many non-motor dysfunctions, including depression, anxiety and olfactory impairments, arise earlier in the course of PD, prior to the onset of the motor dysfunction, and many symptoms, such as those related to cognitive dysfunction, emerge during the late phases of the disease [21, 22, 23].
Given the complexity of the PD syndrome, it is clear that dysfunctions within numerous neurotransmitter systems, including the DAergic, noradrenergic, serotonergic, and cholinergic systems, as well as the formation of Lewy bodies, the neuroinflammatory profile and the discrete patterns of cell death, will all converge to elicit the multifaceted pattern of symptoms that manifest as the disease progresses. Historically, the loss of midbrain substantia nigra (SNc) DA neurons has been strongly associated with the emergence of the motor impairments and changes to non-DA systems has been associated with many cognitive symptoms. However, in both patient populations and in rodent models, more controlled, systematic analysis of the relationship between neurological insult and behaviour has revealed an important role for DA in several cognitive and neuropsychiatric impairments. Moreover, the perception that the DAergic ventral tegmental area (VTA) neurons are relatively spared in PD has been challenged numerous times, with at least 9 independent analyses of post-mortem brain tissue revealing ~53% loss of VTA cells (in brains with ~67% loss of SNc neurons) [24]. Thus, it is reasonable to consider the impact of this notable mesolimbic/mesocortical degeneration on the cognitive features of the disease.
Three parallel cortico-striatal-pallido-thalamo-cortical loops exist, which are modulated by DA input from both the SNc and the VTA (Fig. 1). The mesostriatal DAergic circuit, which comprises the ventral midbrain, posterolateral putamen, dorsolateral subthalamic nucleus, and primary motor cortex, controls motor function, but recent studies also reveal roles in cognitive functions. The mesocortical DAergic circuit, which comprises the head of the caudate nucleus, rostral putamen, intermediate zone of subthalamic nucleus, and the dorsolateral prefrontal cortex, is involved in executive functions. The mesolimbic DAergic circuit, which comprises the nucleus accumbens, ventromedial striatum, rostral ventral, ventromedial subthalamic nucleus and anterior cingulate cortex, is involved in reward processing and apathy/depression.
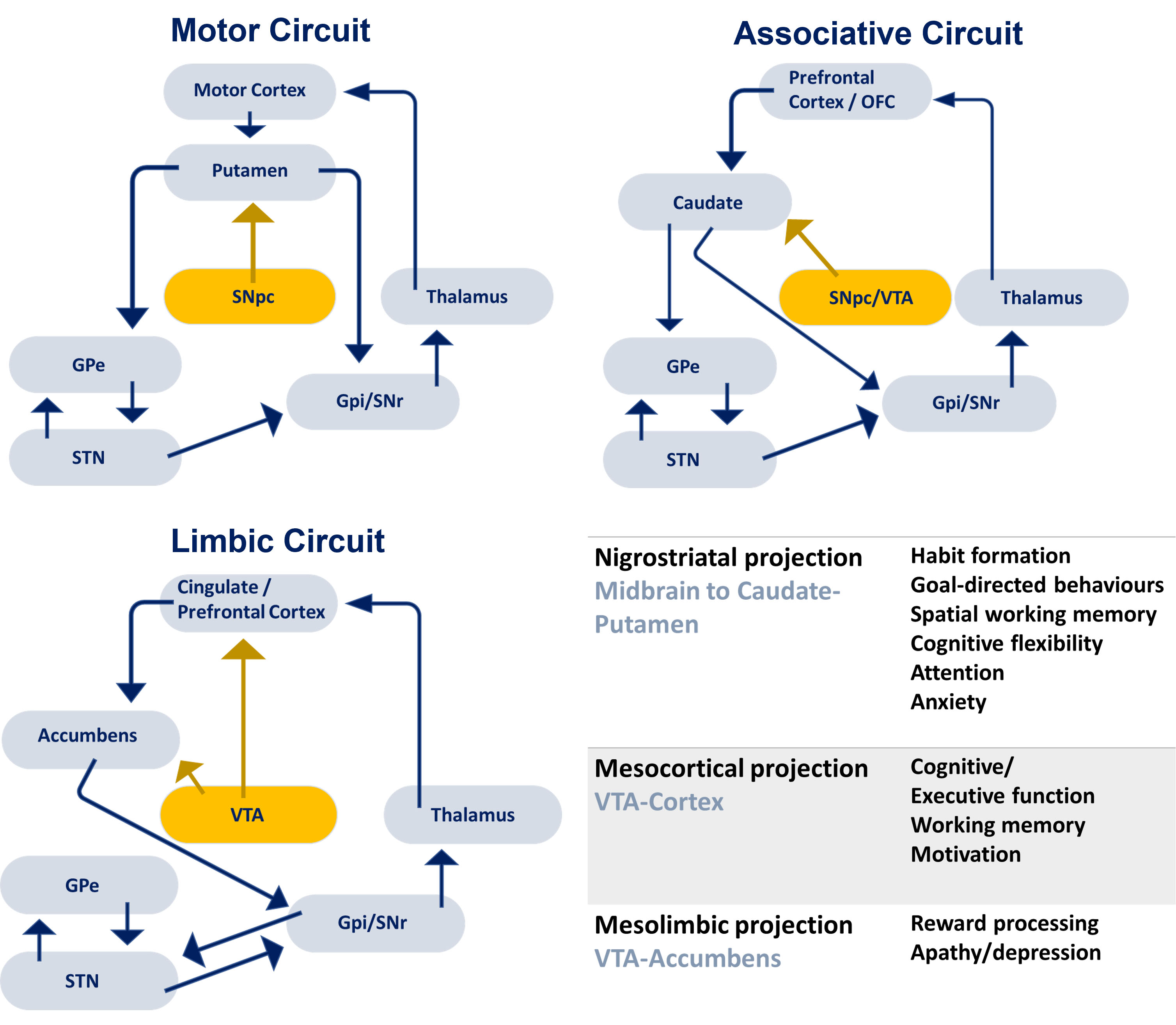 Fig. 1.
Fig. 1.DAergic projections from the SNc and VTA innervate the caudate, putamen, nucleus accumbens and cortex and modulate motor, associative and limbic circuits. Different DA projections have been reported to support a variety of cognitive and neuropsychiatric functions that are affected in PD. SNc, substantia nigra pars compacta; SNr, substantia niga reticulata; VTA, ventral tegmental area; STN, subthalamic nucleus; GPe, globus pallidus external segment; GPi, globus pallidus internal segment; OFC, orbitofrontal cortex.
Evidence from animal models has demonstrated that subregional mesostriatal and mesocortical DA transmission is important for habit formation and goal-directed behaviors [25, 26, 27]. Indeed, in both PD patients and healthy adults, evidence of a role for DA in goal-directed behaviors [28] and stimulus-response (“S-R”) learning [29] has been identified. One hypothesis suggests that the slow and effortful performance of PD patients may reflect a reliance upon goal-directed processing of the environment [30]. In accordance with this, a recent study used a computer typing task to demonstrate disruption of motor habits and a greater reliance on goal-directed behaviours to complete the task [31], although evidence of intact S-R processing and a tendency to rely more upon the habitual response system has also been reported in PD patients [32].
Working memory dysfunction and cognitive dysfunction are frequently reported in PD patients, and indeed, DA has often been implicated in working memory function [33]. For example, impaired spatial working memory has been associated with decreased DA in the nigrostriatal caudate nucleus, but not mesocortical DA transmission [34]. In PD patients, reduced DA in the caudate nucleus correlated with impairments in verbal fluency [35, 36] and verbal working memory [37]. Furthermore, in PD, loss of DA binding in the caudate nucleus has been correlated with the attentional processing, as assessed by the interference task on the Stroop test [38].
DA transmission within mesolimbic pathways has long been associated with motivation and reward processing [39, 40, 41, 42]. Consistent with this, it has been reported that PD patients manifest impairments in reward processing [43, 44] and appetitive motivation [45]. Indeed, aberrant reward processing was related specifically to cognitive inflexibility and DA cell loss in a cohort of non-medicated PD patients [46, 47], suggesting that impairments in reward-related behaviours constitutes an important aspect of the non-motor syndrome. Additionally, it is well accepted that DA agonist treatment can enhance novelty-seeking behaviours and reward learning [43], demonstrating a DAergic basis to this neural process. Interestingly, decreased DA availability in the mesolimbic system and caudate nucleus have also been reported to distinguish PD with dementia from those without dementia [48] and the mesolimibic pathway has been associated with depression in PD [49].
DA replacement improves motor dysfunctions in PD, but several lines of research suggest that improvements in cognition may also occur. Levodopa has been reported to alleviate working memory impairments (although an impairment in attentional set-shifting performance was not attenuated) [50]. DAergic medications can improve response inhibition [51] and spatial working memory deficits, with the latter being associated with prefrontal cortical activation [52]. Interestingly, in a study of reinforcement learning in PD, the pharmacological replacement of DA diminished the emphasis on negative (but not positive) outcomes, which ultimately improved learning behaviours and was associated with activity in the caudate nucleus [53].
Since several non-motor and neuropsychiatric symptoms, such as depression, anxiety, dyspnea, pain, sweating, worsen in the absence of anti-Parkinsonian medications, it has also been suggested that these may have a DAergic basis and may be improved by DA treatments (for a comprehensive review see [54]). For example, a double-blind placebo-controlled trial of dopamine agonist pramipexole over 12 weeks revealed a significant improvement in symptoms of depression, as measured by the 15-item geriatric depression scale and the unified Parkinson’s disease rating scale (part 1 depression item score) in PD patients, suggesting that DA replacement can modulate this impairment [55]. Additionally, a prospective study of deep brain stimulation revealed the emergence of apathy, anxiety and depression when DA treatments were significantly reduced and a correlation between these symptoms and increased raclopride binding along the mesolimbic pathway. This study also provided evidence of improvement in these symptoms with the re-introduction of DA agonists [56].
Thus, whilst a broad range of non-motor symptoms manifest in PD and many of these are likely underpinned by complex non-DAergic pathology, there is evidence that some aspects of cognitive/memory dysfunction are strongly associated with DA transmission and are responsive to DA replacement. Indeed, the relationship between DA loss, behaviour and DA replacement is complex. DA is hypothesised to function as an inverted U, with too little and too much impairing performance, and oral pharmacological treatments will have off-target effects associated with cortical imbalances, which can also manifest as impairments in cognitive function [57, 58, 59, 60]. Moreover, it is well-documented that long-term exposure to DA agonists can lead to impulse control disorders and dopamine dysregulation syndrome [61, 62]. Thus, precise titration of DA is likely required to observe improvements in tests of cognitive, memory or neuropsychiatric function.
Identifying and treating non-motor symptoms in PD patients is confounded by significant variability across patients and the complexity of the disease pathogenesis over time. Importantly, however, preclinical studies that have aimed to elucidate the role of DA transmission in cognitive and neuropsychiatric functions have identified similar relevant pathways to those identified in patients.
Motivation and reward-processing are strongly associated with VTA/mesolimbic DA systems in rodent models, with an abundance of evidence demonstrating that disruption of these mesolimbic pathways impairs diverse aspects of motivational processing [39, 42, 63, 64, 65].
In accordance with theories positing impaired use of habitual S-R associations in patients [30, 32], evidence of a role for both dorsolateral and dorsomedial striatal DAergic transmission in habitual behaviours has been reported [25, 26, 27, 66]. Nigrostriatal projects to the dorsolateral striatum support motor function, but have also been reported to be important in the expression of anxiety [67] and value-based decision making [68]. Dorsomedial projections which emanate largely from the VTA are implicated in cognitive flexibility [69] and anxiety [67]. Furthermore, consistent with patient data, impaired working memory processes have been reported repeatedly in the bilateral 6-OHDA model of PD [70, 71, 72].
Impaired attentional processing in the bilateral intrastriatal 6-OHDA model has been demonstrated using a reaction time task, and improvement in performance was evident after DA agonist treatment, implicating DA in this aspect of performance [73]. Loss of DA impaired memory performance in the novel object recognition task in a bilateral model of PD, and this was improved upon administration of L-dopa [74]. Thus, the evidence from rodent models validates the involvement of DA in distinct cognitive processes and provides further evidence that DA replacement can improve these processes.
Based on the evidence discussed above, it is reasonable to consider to what extent cell and gene ATMPs aiming to replace the lost DA may impact on DA-dependent cognitive and neuropsychiatric symptoms. As discussed above, there is evidence that Levodopa and DA agonists can improve some cognitive, neuropsychiatric and autonomic non-motor symptoms. Pharmacological interventions can, however, induce off-target effects due to their likely impact on cortical function, which makes cell and gene therapies attractive strategies.
It has been reported that mouse-derived [75], rat-derived [76, 77, 78] and human-derived [79] DAergic fetal ventral mesencephalic (VM) grafts can all improve aspects of cognitive performance. These studies all used the unilateral 6-OHDA lesion model, due to the ability to assess cell transplants long-term in the presence of stable DA depletion, and without the neurorecovery observed in more partial PD models [80]. These studies used an operant behavioural task, called the lateralised choice reaction time (LCRT) task, designed to probe motivational processing, visuospatial function and attention. It was reported that human fetal mesencephalic grafts, which harboured both A9 and A10-like DA neurons, ameliorated motor biases (as demonstrated using the drug induced rotation tests), as well as improving all cognitive dysfunctions [79]. Importantly, a systematic analysis of the influence of DAergic, serotonergic and noradrenergic systems on performance on this LCRT task confirmed that these behaviours are driven by DA transmission and are independent of serotonergic and noradrenergic activity [81].
There are very limited data available on the impact of cell or gene ATMPs on cognitive or neuropsychiatric dysfunctions in PD. In one of the earliest transplant clinical trial, promising results were reported after grafting of adrenal medullary tissue in 7 PD patients [82]. They observed amelioration of multiple neuropsychological impairments at 3 months post-graft, including deficits in visuospatial and visuoperceptual processing and memory impairment related to the ability to organise response outputs. However, improvements were not evident in tasks assessing immediate and delayed memory function. Compared to unoperated PD patients and neurologically intact participants, grafted patients performed close to control levels. Additionally, it has been reported that PD patients that received fetal VM tissue transplants demonstrated improvements in verbal memory at 12 and 24 (but not 36) months post-graft, based on the immediate and delayed verbal memory components of the Wechsler Memory Scale [83]. Thus, although there is a lack of evidence demonstrating long term benefit, these early improvements suggested the potential for cell replacement therapies to alleviate cognitive-type dysfunctions.
It has been reported that fetal-derived VM tissue grafts do not improve non-motor dysfunctions in PD patients at 12 months post-transplant [84]. Here, 40 PD patients were enrolled in a double-blind study and neuropsychological analysis of the patients revealed no change in performance on tasks assessing attention, verbal and working memory, abstract reasoning and executive function, visuospatial function, amongst others, at 12 months post-VM DA cell transplant. However, importantly, even analysis of motor function revealed no significant effect of the cell replacement therapy at this stage using the UPDRS, Hoehn & Yahr, and Schwab & England scales. The authors conclude that while the transplantation procedure was safe and not detrimental to the cognitive function of the patients, one year post-surgery may be too soon to observe changes in any behaviours. Interestingly, a subsequent analysis of a subset of the original cohort of patients reported in Trott et al. [84] revealed improved performance on the UPDRS at 2 and 4 years post-transplant, although cognitive function was not reported [85].
In asking whether it is plausible for cell or gene ATMPs to improve cognitive dysfunctions in PD, some things must be considered. Firstly, it is important to scrutinize the nature of the tasks being assessed. There are a wide variety of non-motor dysfunctions and the evidence to data suggests that some, but not all, involve the nigrostriatal or mesolimbic DA systems. Thus, it is critical to focus on tasks that are dependent on DA transmission and likely to benefit from a targeted DA replacement strategy. Here we touch upon motivation, reward processing, working memory and habitual S-R behaviours and demonstrate using clinical and preclinical data that there is a role for DA transmission in these behaviours and that there is some evidence of anti-Parkinsonian drugs influencing these neural processes.
Secondly, the nature of the experimental design makes it more challenging to assess the impact of the cell or gene therapy ATMPs on cognitive dysfunctions. For example, given the permanent nature of the transplants or viral vector infusions, the experimental design would necessarily involve the use of a sham grafted or a natural history control group. This provides an opportunity to identify significant decline in the sham/control cohort to allow room to see improvement or maintenance of performance in the grafted cohort. Moreover, and importantly, post-transplant PD patients typically continue to take oral Levodopa and other anti-Parkinsonian medications, in the presence of the graft, which makes assessing the impact of the graft in isolation more challenging. Removing the influence of oral pharmacotherapeutics becomes especially important given our knowledge of off-target effects from Levodopa and DA agonists and their (sometimes detrimental) impact on cognitive functions [57, 58, 59, 60]. It may be necessary, therefore, to assess the change in performance on behavioural tasks both off- and on-DA medication in order to detect the impact of the therapeutic intervention. Thus, the data available to date are insufficient to demonstrate the impact of DA rich transplants on cognitive function and these challenges in experimental design likely contribute to the discrepancy in data collected between preclinical and clinical studies.
There is a wealth of data to suggest that physiological DA transmission is fundamental to support several cognitive and neuropsychiatric processes, including motivation/reward processing, working memory and habit/goal-directed behaviours. These processes are known to be disrupted in PD patients and there is evidence that oral pharmacotherapeutics influence these behaviours. Preclinical animal models have been successful in demonstrating that DA rich grafts can improve cognitive dysfunctions, but clinical studies are sparse and it is necessary to improve our understanding of the influence of ATMPs on DA-dependent cognitive and neuropsychiatric processes.
Indeed, this may be an opportunity to develop an enhanced cell therapy product capable of ameliorating cognitive and motor symptoms, thereby improving quality of life for the patient. Re-assessment of the current transplantation approaches may need to be considered to achieve this. For example, rather than the current focus on purified A9-like DA cell populations, it may be pertinent to develop a midbrain-like cell therapy product that includes mesolimbic A10-like DA cell populations, which would more closely mimic the original fetal-derived cell grafts. Or the ectopic transplant strategy may be replaced by reinvigorated interest in midbrain transplantation, in order to achieve more complete, biologically relevant circuitry reconstruction. Thus, in conclusion, it is critical to (1) consider this opportunity to derive greater knowledge of the biological mechanisms supporting DA-dependent cognitive dysfunctions and (2) harness opportunities to develop an enhanced cell therapy product capable of ameliorating a wider range of dysfunctions.
ML conceived of and wrote the manuscript.
Not applicable.
Not applicable.
ML is supported by a Parkinson’s UK Senior Research Fellowship F1502.
The author declares no conflict of interest.