1. Introduction
Absence seizures are characterized by periods of behavioral arrest and amnesia
without overt convulsions [1]. In patients, these generalized seizures typically
last 2–15 seconds with corresponding bilateral, synchronous ~3
Hz electrographic spike-and-wave discharges (SWDs) [2]. The generalized nature of
absences, however, should not be interpreted to mean that there is no specific
focus or generating region.
Current first-line medications to control absence seizures associate this
disorder with pathological thalamic relay neuron activity [3]. Ethosuximide and
valproic acid are two commonly prescribed medications to treat absence seizures,
with approximately equal efficacy (~55%) [4, 5]. Both agents act
as T-type Ca channel blockers, thus directly inhibiting
Ca-dependent plateau potentials and bursting in thalamic relay neurons
[6]. Cope et al. [7] demonstrated that the propensity of thalamic relay
neurons to express T-type Ca channel-mediated bursts is positively
correlated with the level of -aminobutyric acid type A-associated
(GABAergic) tonic inhibition. Several rodent models of absence epilepsy display
aberrantly enhanced tonic inhibition in thalamic relay neurons [8, 9, 10], leading
to the conclusion that enhanced thalamic tonic inhibition is “necessary and
sufficient for the generation of typical absence seizures” [9]. However, it
remains to be understood why current first-line medications have limited efficacy
in controlling human absence epilepsy.
Absence epilepsy is a thalamocortical disorder, thus a “cortical focus theory”
for absence seizures has also been proposed in many absence animal models
[11, 12, 13, 14, 15]. The cortical focus theory is rooted in findings that SWD initiation was
localized to the perioral region of the somatosensory cortex in multiple animal
models of absence epilepsy [11, 14, 16, 17, 18, 19]. Local injection of certain drugs into
this area was enough to suppress absence seizure expression [20, 21, 22, 23]. Direct
administration of the endogenous neurosteroid allopregnanolone (ALLO), or its
synthetic derivative ganaxolone (GANX), into the primary somatosensory cortex of
SWD-expressing Wistar Albino Glaxo Rijswijk (WAG/Rij) rats reduced both the
number and duration of SWDs [20].
ALLO and GANX are positive allosteric modulators of -aminobutyric acid type-A (GABA) receptors, having
their largest effects at subunit-containing GABA receptors and
being relatively selective for this subtype at low concentrations [24]. GANX has
been evaluated as an antiepileptic drug in humans [25, 26, 27] for the treatment of
infantile spasms [28] and has shown efficacy with minimal side effects as a
treatment for catamenial epilepsy [24] and partial seizures [29]. In animal
studies of partial seizures, subcutaneous administration of GANX displayed
antiseizure potential even at relatively low doses (3 mg/kg) [30]. However, in
two animal models of absence epilepsy (pentylenetetrazole, PTZ;
gamma-hydroxybutyric acid, GHB) GANX exacerbated absence seizures and even
produced SWDs in wild-type rats when either systemically administered at
20 mg/kg [31] or administered directly into the ventrobasal thalamus
[20]. These findings might be explained if high GANX concentrations stimulated
thalamic tonic inhibition via subunit-containing GABA receptors,
as observed in previous studies [8, 9, 10]. These findings suggest a dichotomy of
effects for neurosteroids regarding absence epilepsy: direct administration into
the somatosensory cortex decreases SWD generation and duration, whereas
systemic administration of high concentrations results in sedation and SWD
exacerbation or generation.
This heritable epilepsy in humans has been traced to an arginine-to-glutamine
substitution at position 43 of the GABA receptor 2 subunit
(2R43Q) that confers a variety of phenotypes, the most common being
Childhood Absence Epilepsy (CAE) and febrile seizures [32]. Patients expressing
this mutation show evidence of a hyperexcitable cortex and diminished
intracortical inhibition [33] that is believed to contribute to SWDs [34].
Patients affected with the 2R43Q mutation are all heterozygous.
Heterozygous knock-in (RQ) mice display absence-like seizures, generalized SWDs
and an early developmental onset of seizure susceptibility, all consistent with
observations from affected human families [35]. RQ mouse cortical neurons also
display a higher spontaneous-firing rate [36].
In this study we used continuous video- electroencephalogram (EEG) monitoring
and voltage-clamp electrophysiology to show that RQ mice that display SWDs also
lack tonic inhibition in both cortical layer 2/3 pyramidal and thalamic
relay neurons. We then used selective pharmacology to modulate cortical tonic
inhibition in wild-type mice, introducing a novel model of absence epilepsy
employing a previously unexplored mechanism in which decreased cortical
tonic inhibition is sufficient to trigger SWDs. Finally, we suppressed the
expression and duration of SWDs in absence (RQ) mice by rescuing the
lost cortical tonic inhibition via low-dose systemic GANX administration.
Together with previous findings, our data suggest that an optimal level of tonic
inhibition must be maintained throughout the thalamocortical circuit to ensure
normal function and offer a new therapeutic option (low-dose GANX) for patients
where SWDs are caused by alternative mechanisms.
2. Methods
2.1 EEG Implantation and Monitoring of SWDs
Our study used male and female wild-type Harlan C57BL/6J-OlaHsd and
2R43Q knock-in mice bred into a background of Harlan C57BL/6J-OlaHsd
mice. Behavioral and electrographic markers of absence epilepsy in these animals
were confirmed by video-EEG monitoring. Surgery and electrode implantation were
performed as described by Nelson et al. [37]. Briefly, P24 mice were
implanted under isoflurane (#sc-363629Rx; Santa Crus Biotechnology, Dallas, TX,
USA) anesthesia (1%–2% in 100% O) for chronic EEG recordings with gold
plated miniature screw electrodes over the right and left frontal and parietal
cortices, and one over the cerebellum as a reference. Two vinyl-coated braided
stainless steel wire electrodes were placed in the nuchal muscle for
electromyogram (EMG) recording of muscle activity. All electrodes were gathered
into a flexible cable and connected to the Multichannel Neurophysiology Recording
system (Tucker-Davis Technologies, TDT, Alachua, FL, USA). EEG and EMG signals
were collected continuously at a sampling rate of 256 Hz (digitally filtered
between 0.1 and 100 Hz). Continuous EEG recordings with occasional video
monitoring were made and SWDs were manually scored off-line. Animals were given a
3-day recovery period after surgery before initiating SWD-scoring. A SWD event
was defined as a brief (~2 seconds long) ~6 Hz
signal synchronized across all EEG leads, with a corresponding lack of signal in
the EMG lead. Only SWD events that occurred 2 min from slow-wave-sleep periods
were used for quantification. SWD event durations were measured from the first
synchronized positive peak signal to the last synchronized positive peak within
an event. “Seizures” were defined as groups of SWD events separated from other
events by 30 seconds. Ictal intervals were defined as the time between the
beginnings of consecutive seizures. All animal procedures followed the National
Institutes of Health Guide for the Care and Use of Laboratory Animals and were
approved by the Institutional Animal Care and Use Committee (IACUC) of the
University of Wisconsin-Madison (No. A3368-01). All facilities were inspected and
accredited by the Association for Assessment and Accreditation of Laboratory
Animal Care International (AAALAC).
2.2 Drugs and Injection Schedule
L655,708 (L9787), GANX (G7795) and THIP (T101;
4,5,6,7-tetrahydroisoxazolo[5,4-c]-pyridine-3-ol HCl) were all obtained from
Sigma (St. Louis, MO, USA). L655 and GANX were dissolved in a 30% dimethyl sulfoxide (DMSO)-saline
solution (v/v), whereas THIP was dissolved in 100% saline. Mice were
intraperitoneally (i.p.) injected with 2 mg/kg doses of L655, 2 and 5 mg/kg doses
of GANX, or 0.5 and 1.5 mg/kg doses of THIP. 160 µL of solution was
injected for each drug. L655 was administered to wild type (RR) mice 2 and 4
hours after lights out for 2 consecutive days beginning 5 days after surgery.
These mice were not injected for the subsequent 2 days but were given vehicle
injections on day 9. GANX or THIP injections were administered to RQ mice 1 and 4
hours after lights out. Drug injections for RQ mice began on day 5 post surgery
and consisted of 2 injections of one drug and dose, with a different drug and
dose for days 6, 10, and 11. No injections were given to RQ mice on days 7–9.
2.3 Whole-Cell Patch Clamp Experiments
Horizontal slices (400 µm) were prepared from the brains of RR and
RQ mice of either sex (16–26 days old). All procedures were approved by the
University of Wisconsin IACUC. Mice were anesthetized with isoflurane,
decapitated, and the brain was removed and placed in an ice-cold cutting solution
containing (in mM): 125 NaCl, 25 NaHCO, 2.5 KCl, 1.25 NaHPO, 0.5 CaCl, 3.35
MgCl, 25 D-Glucose, 13.87 sucrose, and bubbled with 95% O and 5% CO. Slices
were cut using a vibratome (Leica VT 1000S, Global Medical Imaging; Ramsey, MN,
USA) and placed in a bubbled incubation chamber containing standard artificial
cerebrospinal fluid (aCSF) (in mM): 125 NaCl, 25 NaHCO, 2.5 KCl, 1.25 NaHPO, 2
CaCl, 1 MgCl, 25 D-Glucose, at room temperature for 1 hour before being used
for recordings. Whole cell patch-clamp recordings were made from somatosensory
cortical layer 2/3 pyramidal cells or ventrobasal thalamic relay cells,
visualized using an upright differential interference contrast microscope
(Axioskop FS2, Zeiss; Oberkochen, Germany). Patch pipettes were pulled from
thin-walled borosilicate glass (World Precision Instruments; Sarasota, FL, USA)
with a resistance of 3–5 M when filled with an intracellular solution
containing (in mM): 140 KCl, 10 EGTA, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 20 phosphocreatine, 2 Mg2ATP, 0.3
NaGTP (pH 7.3, 310 mOsm). Voltage clamp (–60 mV) recordings were made in a
submerged chamber, perfused with bubbled aCSF (4 mL/min) containing (500 nM)
tetrodotoxin, (25 µM) DNQX, and (50 µM) AP5 at room
temperature using a MultiClamp 700B amplifier (Axon Instruments; Foster City, CA,
USA), filtered at 4 kHz and digitized at 10 kHz using a Digidata 1322A
analog-digital interface (Axon Instruments). Data were acquired to a Macintosh G4
(Apple Computer; Cupertino, CA, USA) using Axograph X v1.1.4 (Molecular Devices;
Sunnyvale, CA, USA).
Data segments (30 seconds) just prior to and 90 seconds after drug
administration were analyzed to quantify inhibitory tonic currents. All-point
amplitude histograms were computed for each segment and fit with a Gaussian
function only to the outward current portions relative to the peak in order to
omit components arising from inward miniature inhibitory post-synaptic currents
(mIPSCs) [38]. Tonic current was calculated as the difference between the fitted
Gaussian means before and after (100 nM or 1 µM) THIP, (30 nM) ALLO or
(10 nM) GANX administration. Current density (pA/pF) was calculated by dividing
the current by cell capacitance. Bicuculline (100 µM; bicuculline
methiodide #2503, Tocris Bioscience, Minneapolis, MN, USA)) was added at the
conclusion of some experiments for each drug tested to verify full current block
and, therefore, only GABAergic contribution.
2.4 Statistics
The Kruskal-Wallis test of medians was used to compare multiple groups with a
Dunn’s post-hoc evaluation. Tonic current amplitude and density data were
normally distributed; therefore, an analysis of variance (ANOVA) was used to
compare multiple groups with a Bonferroni post-hoc evaluation. A p-value
of 0.05 is considered significant. MATLAB (version 2013b, Mathworks Inc,
Natick, MA, USA) and Prism (macOS v10.0.0, GraphPad Software, Boston, MA, USA)
software were used.
3. Results
3.1 RQ Mice Express SWDs and Absence Epilepsy
The 2R43Q mutation confers absence seizures and generalized SWDs in
humans [32] and knock-in (RQ) mice [35], consistent with results presented in
this study. Fig. 1 illustrates bilateral, synchronous (~6 Hz)
SWDs in a RQ mouse using continuous EEG and EMG recordings. Quantification was
done off-line after recordings were completed. A “seizure” was classified as
two or more individual SWD events occurring 30 seconds apart. SWDs were
assessed for individual event duration, events per seizure, seizure duration and
ictal intervals. EEG and EMG recordings from one RQ mouse during a seizure are
presented (Fig. 1a,b), along with quantified SWD assessment for three different
RQ mice (Fig. 1c). All RQ mice assessed with EEG and EMG monitoring presented
with synchronized SWDs across all EEG leads with coincident cessation of EMG
activity. Some spindle activity but no SWDs were observed in drug-naïve wild
type (RR) mice.
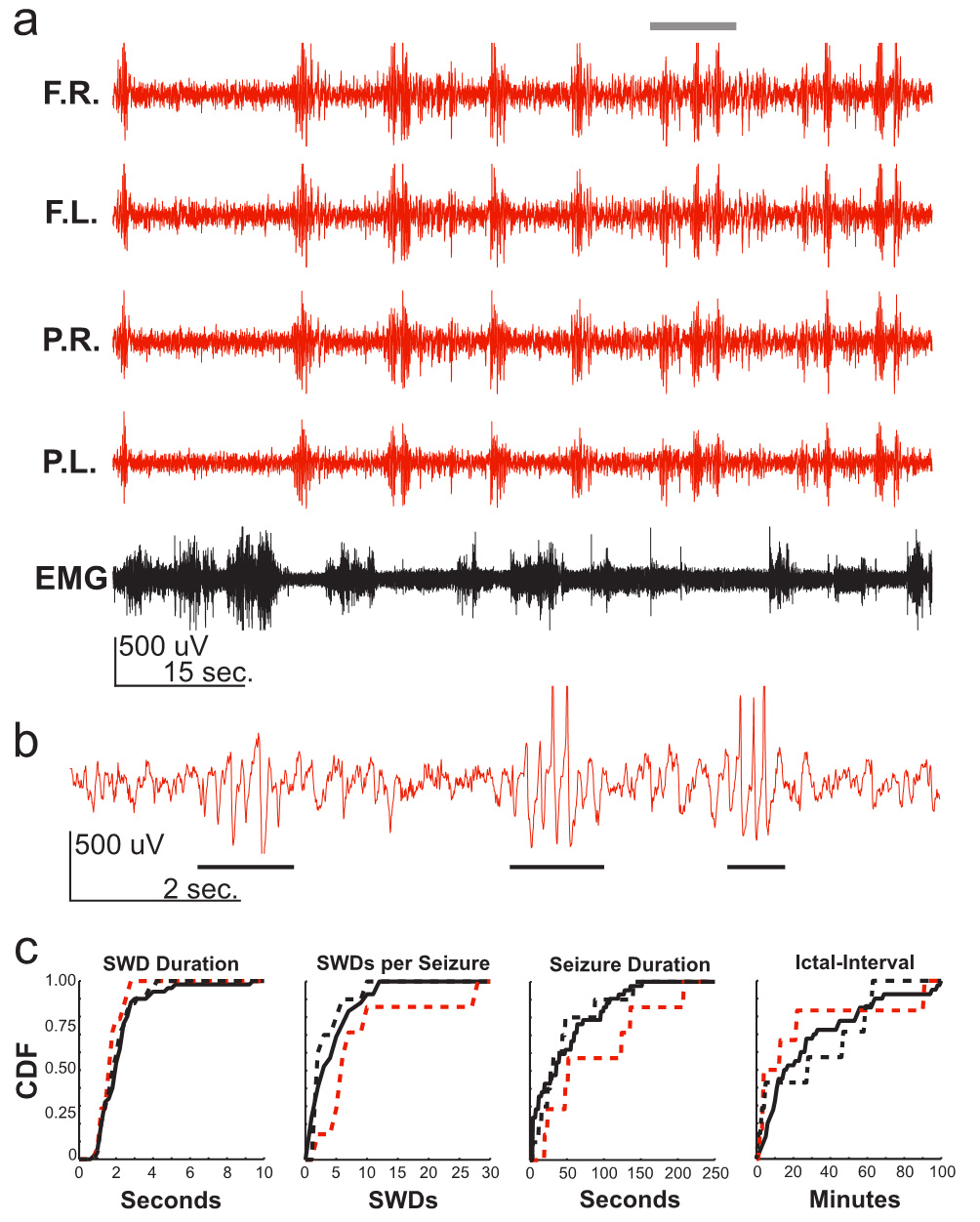 Fig. 1.
Fig. 1.
RQ mice express spike-and-wave discharges (SWDs) associated with
absence epilepsy. (a) Electroencephalogram (EEG) recording of an RQ mouse (red).
Top trace to bottom trace: frontal right cortex (F.R.); frontal left cortex
(F.L.); parietal right cortex (P.R.); parietal left cortex (P.L.); electromyogram
(EMG). Note the brief yet frequent (~11 times during the
1.5-minute segment) synchronized events that occur across all EEG leads during
the absence of signal in the EMG. (b) Expanded F.R. EEG recording from grey bar
in (a) (10 seconds). Note the brief ~6 Hz SWD events (black bars)
that occur 3 times during the 10 second trace. (c) Cumulative probability
distributions from three different RQ mice (solid & dashed black lines and red
dashed line represent individual mice) all display similar characteristics for
SWD event durations, SWDs per seizure, seizure durations and ictal-intervals.
SWDs were not detected in litter-mate control mice (not shown). CDF, cumulative density function;
RQ, R43Q mutant.
3.2 GABAergic Tonic Inhibition Is Abolished in RQ Cortical and
Thalamic Principal Neurons
Although RQ mice express slightly decreased IPSC amplitudes [35], this mutation
has also been shown to hinder GABA receptor assembly, trafficking and
surface expression [39, 40, 41, 42, 43]. Based on studies in transfected cultured
neurons, Eugène et al. [42] reported that this mutation may
contribute to absence epilepsy by reducing tonic inhibition. To directly test
this hypothesis in RQ animals, we examined tonic inhibition levels in brain
slices from RR and RQ knock-in mice. Using whole cell voltage clamp recordings,
we found that whereas RR neurons exhibit a substantial inhibitory tonic current,
this current was entirely abolished in RQ mice somatosensory cortical layer 2/3
neurons (mean standard error of the mean (SEM) in pA, N) (RR: 6.0 0.8, 4; RQ: –1.2
1.6, 4, p 0.05; Fig. 2a,b), as well as in thalamic relay neurons
(RR: 6.9 2.2, 9; RQ: 0.6 0.7, 6, p 0.05; Fig. 2c,d).
One-sample t-test indicated that tonic currents in RQ were
indistinguishable from zero (p 0.45).
 Fig. 2.
Fig. 2.
Tonic currents are abolished in RQ cortex and thalamus. (a)
Example voltage-clamp traces for RR (black) and RQ (red) cortical layer 2/3 cell
recordings during 100 µM Bicuculline administration (maroon bars).
Insets; Corresponding all-points amplitude histograms for data before (black) and
after (grey) bicuculline administration. Histograms were fit with a Gaussian
function (dark grey and maroon traces) only on the right side of the
distribution, thus omitting components due to phasic miniature inhibitory
post-synaptic currents (mIPSCs). (b) Tonic current amplitude (pA) (left axis) and
tonic current density (pA/pF) (right axis) are abolished in RQ cortical cells
(*p 0.05) compared to control. (c,d) Same as a-b, but for
ventrobasal thalamic relay neurons. RR, wild type.
3.3 GABA Receptor Function or Expression Is Altered in a
Region-Specific Manner
The tonic current in RR somatosensory cortical layer 2/3 cells (Fig. 2a) was
completely blocked by the 5 subunit-selective inverse agonist L655,708
(L655: 30 µM; Fig. 3a), consistent with previous studies showing
this subunit is responsible for most or all of the native tonic inhibition in
these neurons [44].
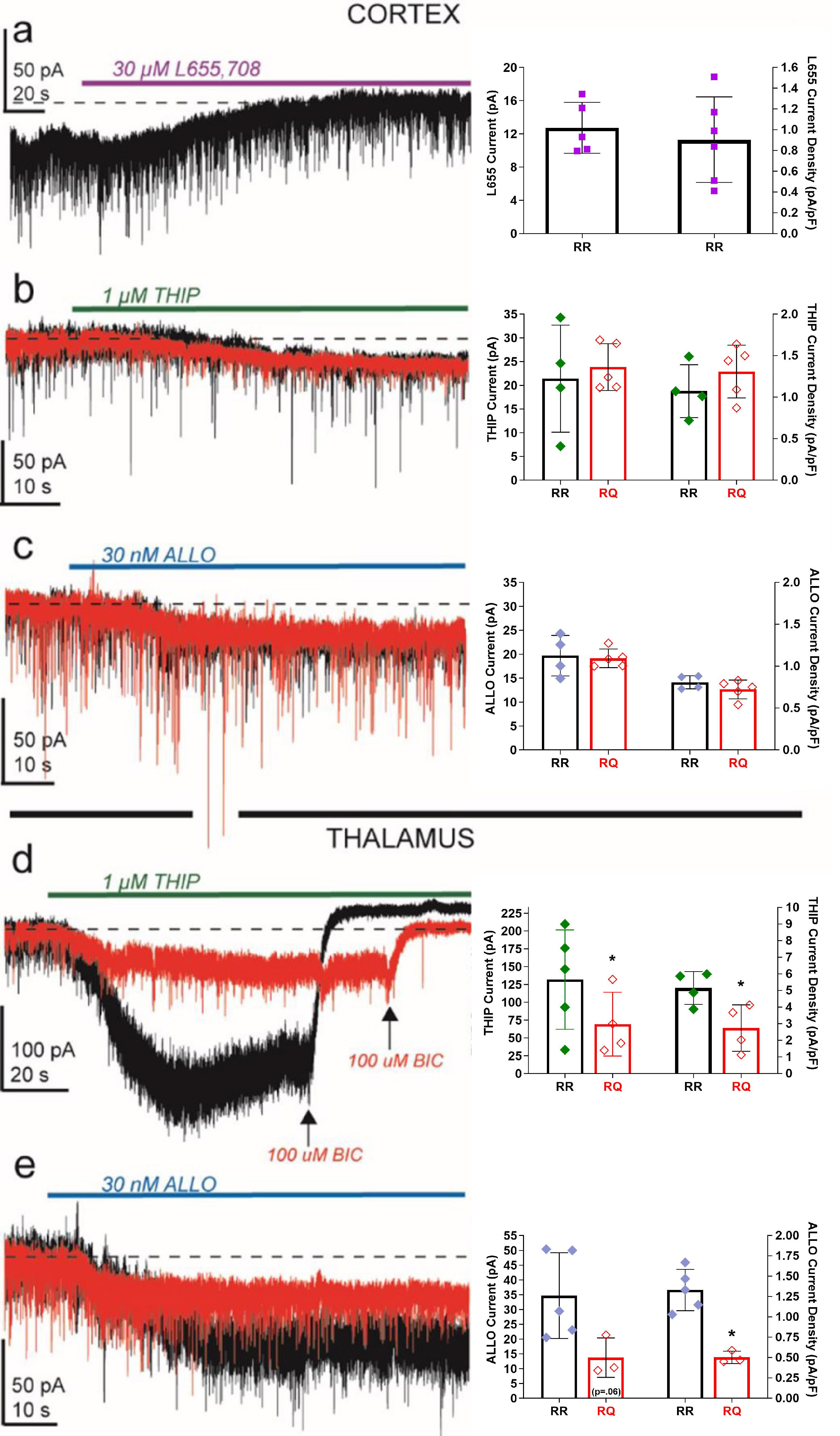 Fig. 3.
Fig. 3.
RQ mice display region- and subunit-specific changes in tonic
inhibition. (a) Example voltage-clamp traces for RR cortical layer 2/3 cell
recordings during 30 µM L655,708 administration (purple bar). The
current density blocked by L655,708 was not significantly different than that
blocked by bicuculline (see Fig. 2). In cortical neurons, both THIP. (b) (1
µM; green bar) and allopregnanolone. (c) (ALLO; 30 nM; blue bar)
induce indistinguishable current amplitude and density in RQ (red traces)
compared to RR (black traces). In thalamic relay neurons, however, THIP (d) and
ALLO-induced (e) current densities are significantly reduced in RQ compared to RR
(~50%; *p 0.05). ALLO, allopregnanolone; THIP, 4,5,6,7-tetrahydroisoxazolo[5,4-c]-pyridine-3-ol HCl.
In contrast, application of the agonist THIP (1 µM, a concentration
previously shown to be selective for subunit-containing receptors
[45]) evoked currents of similar magnitude in RR and RQ cortical neurons (mean
SEM in pA, N) (RR: 21.4 5.7, 4; RQ: 23.8 2.2, 5;
p = 0.67; Fig. 3b). A similar profile of effect was observed with
allopregnanolone (ALLO: 30 nM; Fig. 3c), a neurosteroid that also selectively
activates subunit-containing GABA receptors [46, 47]. Together,
these results suggest that receptors containing the subunit are
present in cortical neurons and can be recruited by both exogenous drugs and
endogenous modulators, providing potential pharmacological pathways to rescue
cortical tonic inhibition in cases where it has been genetically compromised.
Distinct from cortical layer 2/3 neurons, thalamic relay neurons rely solely on
subunit-containing GABA receptors to produce inhibitory tonic
currents [7, 45, 48]. We found that RQ relay neurons in the ventrobasal thalamus
responded to THIP (1 µM) with 47% of the current produced in RR
thalamic neurons (mean SEM in pA, N) (RR: 131.7 31.2, 5; RQ: 69.3
22.4, 4, p 0.05; Fig. 3d). Similarly, in RQ thalamic
neurons, ALLO (30 nM) produced 39% of the current observed in RR (RR: 34.7
6.5, 5; RQ: 13.7 3.8, 3, p 0.05; Fig. 3e). These
results suggest that RQ mouse thalamic relay neurons express reduced levels
and/or activation of subunit-containing GABA receptors.
3.4 Blocking Cortical Tonic Inhibition in Wild-Type Mice Produces
SWDs
Previous work has demonstrated that a positive correlation between SWDs and
thalamic inhibitory tonic current amplitude exists [7], leading to the conclusion
that enhanced GABAergic tonic inhibition is “necessary and sufficient” to cause
typical absence epilepsy [9, 10]. In this study, we found (by using the
5 subunit-selective blocker L655) that altering thalamic tonic
inhibition is not “necessary” for SWD generation and, furthermore, that the
loss of 5 subunit-mediated tonic inhibition (present mainly in cortex
and hippocampus) is “sufficient” to produce SWDs (Fig. 4). Inhibitory tonic
currents in somatosensory cortical layer 2/3 principal neurons are generated by
5 subunit-containing GABA receptors, evident by the total block
of this current by the 5 subunit-selective inverse agonist L655 (Fig. 3a). This is in contrast to thalamic relay neurons that do not express a5
subunit-containing GABA receptors [49, 50]. Intraperitoneal (i.p.)
administration of L655, at a concentration (2 mg/kg) known to bind the majority
of 5 subunit-containing receptors [51], produced SWDs
(~6 Hz) in RR mice (RRL6) that are electrographically similar,
yet distinct, to SWDs observed in RQ mice (Fig. 4a,b). L655 administration
induced hallmark synchronous and bilateral SWDs accompanied by behavioral arrest,
although these L655-induced SWDs (L6-SWDs) display longer event durations
(p 0.05), fewer events per seizure (p 0.05) and
shortened seizure durations (p 0.01) compared to RQ (Fig. 4c). The
appearance of SWDs 3-days post final L655-injection (Fig. 4d, hour 1 vehicle)
highlight lingering plasticity and epileptogenesis induced by the earlier acute
insults that provoked SWDs.
 Fig. 4.
Fig. 4.
Blocking cortical tonic inhibition produces SWDs in wild-type
mice. (a) Electroencephalogram (EEG) recording of a wild-type (RR) mouse i.p.
injected with 2 mg/kg of the GABA receptor 5 subunit-selective
inverse agonist L655,708 (RRL6; purple). Similarly to RQ mice, note the brief yet
frequent (~6 times during the 1.5 min. trace) synchronized events
that occur across all EEG leads during the absence of signal in the EMG. (b)
Expanded F.R. EEG recording from grey bar in (a) (10 seconds) displays prolonged
~6 Hz SWD event (black bar). (c) Cumulative distributions show
RRL6 mice (purple line) display significantly longer SWD event durations
(*p 0.05), fewer SWDs per seizure (*p 0.05) and shorter
seizure durations (*p 0.05) than RQ mice (red line). (d)
Quantification of SWDs show that RRL6 mice did not display SWDs (purple boxes)
prior to L655 injection (Hour 1: L655, middle purple bars) but do display SWDs
for hours following injection (Hour 2: *p 0.05; Hour 4: *p 0.05). Black outlined bars represent median values. SWDs were still present
in RRL6 mice 3 days after the final L655 dosing (vehicle: Hour 1, right turquois
diamonds; *p 0.05).
3.5 Rescuing Cortical Tonic Inhibition Attenuates SWDs in RQ Mice
Although RQ principal cortical neurons lack inhibitory tonic currents (Fig. 2a),
these neurons also display an inhibitory conductance in response to selective
subunit-selective GABA receptor agonists THIP (1
µM) and ALLO (30 nm) (Fig. 3b,c). This finding is consistent with
the presence of latent subunit-containing GABA receptors in RQ
cortical neurons and suggests that the lost tonic inhibition in these neurons can
be rescued. We used whole-cell patch-clamp recordings to titrate a concentration
of selective- subunit-containing GABA receptor modulators that
rescued wild-type tonic inhibition levels in RQ cortical neurons. We found (Fig. 5a) that a low concentration of THIP (100 nM) or the synthetic neurosteroid GANX
(10 nM) can activate a latent inhibitory conductance in RQ cortical neurons equal
to the inhibitory tonic current observed in RR cortical neurons.
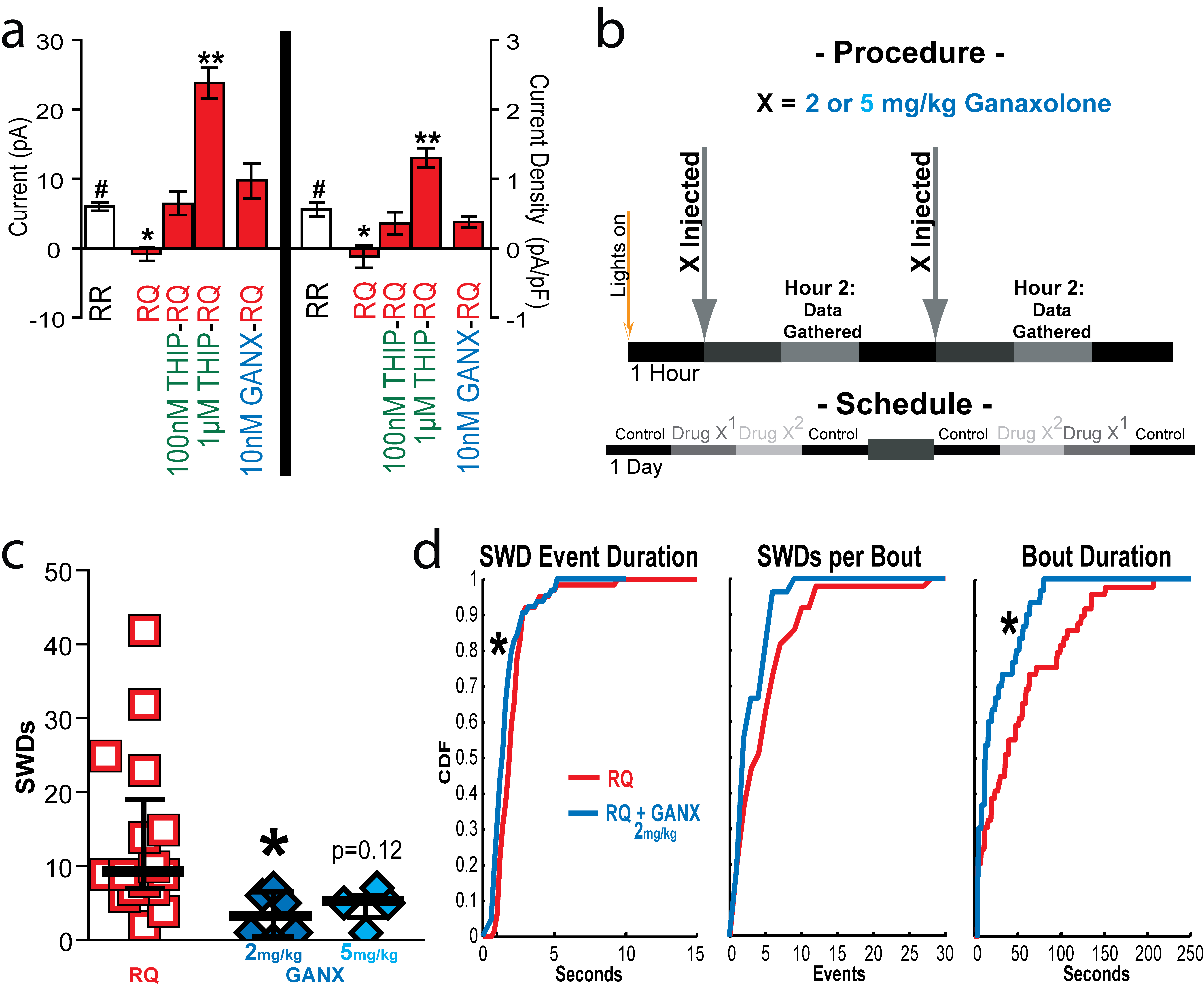 Fig. 5.
Fig. 5.
Rescuing cortical tonic inhibition alleviates SWDs in RQ mice.
(a) Voltage-clamp experiments reveal that tonic current amplitude (left y-axis)
and density (right y-axis) levels can be rescued in RQ (red bars) cortical layer
2/3 neurons with 100 nM THIP or 10 nM ganaxolone (GANX) treatment, whereas a 1
µM THIP produces 2–4 times more holding current amplitude
(**p 0.01) and density (**p 0.01) than that observed in
untreated RR neurons (#). (b) Schematic depicts administration and data
collection (Hour 2) times and drug-day schedules investigating treatment
conditions for RQ mice. GANX (2 and 5 mg/kg) solutions were i.p. injected into RQ
mice twice a day for 4 out of 7 days. (c) RQ-SWD event quantification shows that
the 2 mg/kg GANX (*p 0.05) (Dark Blue) treatment decreased SWD
expression compared to control hours, while 5 mg/kg GANX (p = 0.12)
(Light Blue) treatment trends towards ameliorating SWD expression. (d) Cumulative
distributions of RQ-SWD activity shows that SWD event duration (*p
0.05) and seizure duration (*p 0.05) were decreased with the 2 mg/kg
GANX treatment.
Using video-EEG monitoring, we investigated if treatment with the
subunit-selective GABA receptor agonist (GANX) could ameliorate the SWDs
observed in RQ mice. RQ mice were i.p. injected twice a day with GANX for 4 out
of 7 days (Fig. 5b). Two concentrations of GANX (2 and 5 mg/kg) were tested for
amelioration of SWD expression. We found that only the lower concentration (2
mg/kg) of GANX significantly (p 0.05) decreased RQ-SWD expression
(Fig. 5c). This low dose GANX treatment (2 mg/kg) also decreased seizure duration
(p 0.05) and event duration (p 0.001) (Fig. 5d). The
efficacy of the low GANX dose (2 mg/kg), being half the ED dose that
protects against partial seizures [30, 51, 52], suggests that the mechanism
diminishing SWDs in RQ mice involves activation of latent
subunit-containing GABA receptors in cortical neurons.
4. Discussion
The major findings from this study are that the loss (RQ: Fig. 2) or decrease
(RRL6: Fig. 4) of cortical tonic inhibition accompanies a SWD-expressing
phenotype and that pharmacological replacement of cortical tonic inhibition
(RQ-GANX: Fig. 5c) decreases SWD expression. These findings are consistent with
the conclusion that cortical tonic inhibition levels inversely regulate SWD
expression. Therefore, the causal link between absence epilepsy and inhibitory
tonic currents is at the least bidirectional: increased thalamic tonic
inhibition [7] or decreased cortical tonic inhibition can both lead to
epileptiform activity.
The link between absence seizures and increased subunit-associated
GABA receptor activation in thalamic relay neurons is well established
[8, 9, 10]. The current leading hypothesis from this evidence is that persistent
hyperpolarization of thalamic relay neurons favors T-type Ca channel
availability [7, 45], making these neurons more susceptible to rhythmic bursting
and insensitive to sensory input, considered to be a necessary condition for SWD
generation [9, 10]. Consistent with this hypothesis, ethosuximide and valproic
acid, two different T-type Ca channel blockers, decrease thalamic relay
bursting and are currently the main treatment options to treat absence epilepsy.
However, the efficacy of either drug for this condition is at only
~55% [4]. The evidence presented in this study suggests a second
classification of absence seizure etiology, separate from altered thalamic
activity, that likely accounts for at least a portion of the remaining
~45% of patients that are currently non-responsive to the main
treatment options.
Our findings also suggest that SWDs are not linked to a specific GABA
receptor subtype (5 or ), but rather linked to cortical tonic
inhibitory tone. Rescuing cortical tonic inhibition in RQ mice via activation of
-subunit-associated GABA receptors with GANX, and the subsequent
decrease in SWD expression (Fig. 5) indicates that SWD expression can be
regulated by -subunit-associated tonic inhibition. Additionally, the
selective decrease/block of 5 subunit-associated inhibition (RRL6),
which results in SWD expression (Fig. 4) indicates that SWDs can also be
regulated by 5 subunit-associated tonic inhibition levels.
Collectively, these results provide good evidence that the gating control of SWD
expression is not necessarily linked to any specific GABA receptor subtype
but rather to the general level of cortical tonic inhibitory tone.
Optimal Tonic Inhibition
Studies suggest a dichotomy of effects for neurosteroids in absence epilepsy:
lower levels can ameliorate SWDs in RQ mice, whereas higher concentrations can
result in SWD exacerbation or generation [20]. Based on the results from this
study, we suggest a concentration-dependent relationship of thalamocortical tonic
inhibition in regard to SWDs and absence seizure modulation. Evaluation of
genetic (Genetic Absence Epilepsy Rat from Strasbourg (GAERS), stargazer, lethargic) and pharmaco-induced (GHB, PTZ) rodent
models of absence epilepsy provide ample evidence that excessive thalamic tonic
inhibition triggers SWDs [7, 31]. However, the novel RRL6-absence animal model
introduced here, and the beneficial effects of low-dose GANX treatment in RQ
mice, combine to indicate that a reduction in cortical tonic inhibition also
results in SWDs. These findings suggest that an ‘optimal level’ of tonic
inhibition in the thalamocortical circuit is a requirement for normal function
and that deviation either above or below this optimal range results in aberrant
thalamocortical function, SWDs and absence seizures.
5. Conclusion
Transcranial magnetic stimulation (TMS) studies of human patients harboring the
2R43Q mutation display evidence of a hyperexcitable cortex, increased
intracortical excitability and facilitation, and a decreased intracortical
inhibition [33]. This evidence is in-line with the conclusion that these patients
display a reduced expression of cortical tonic inhibition and that low-dose
ganaxolone treatments might be beneficial in helping control seizure outbreaks.
Although the number of individuals harboring the 2R43Q mutation makes
up only a small percentage of all those that suffer absence seizures, there are
recent findings that provide optimism that a larger portion of the general
absence population would respond positively to this same low-dose ganaxolone
treatment. One recent study employed a thalamocortical computational model that
was optimized via neuronal dynamics captured and measured during SWD events
observed in polygenic (GAERS) or pharmaco-induced (GHB) absence seizure animals.
This investigation concluded that the synchronous, seizure-perpetuating output of
thalamic relay neurons during SWDs is not governed by intrinsic T-type Ca
channel bursting behaviors but rather by the excitability and top-down driving
power of cortical pyramidal neurons [15]. This discovery is significant because
it shifts the regional control of SWD generation and expression into the cortex
for two well-studied absence rodent models (GAERS, GHB) previously regarded as
primary evidence that enhanced thalamic tonic inhibition is the “necessary and
sufficient” hallmark of typical absence epilepsy [9]. Although it has yet to be
investigated, it is possible that a low-dose ganaxolone treatment would
ameliorate the SWDs observed within these two absence rodent models, as well as
within human patients with absence epilepsy that are refractory to the current
first-line medications.
Availability of Data and Materials
All data points generated or analyzed during this study are included in this
article and there are no further underlying data necessary to reproduce the
results.
Author Contributions
KPM and MVJ jointly conceived and designed all experiments for this study with
guidance from ABN, SP and CC. SP developed and provided the RQ mouse model. KPM
and ABN performed experiments. KPM, MVJ, ABN and CC analyzed data. KPM and MVJ
wrote the manuscript. All authors contributed to editorial changes in the
manuscript. All authors read and approved the final manuscript. All authors have
participated sufficiently in the work and agreed to be accountable for all
aspects of the work.
Ethics Approval and Consent to Participate
All animal procedures followed the National Institutes of Health Guide for the
Care and Use of Laboratory Animals and were approved (Animal Welfare Assurance
No. A3368-01) by the IACUC of the University of Wisconsin-Madison. Facilities
were inspected and accredited by AAALAC. We confirm that we have read the
Journal’s position on issues involved in ethical publication and affirm that this
report is consistent with those guidelines.
Acknowledgment
We thank Cynthia Czajkowski, Laura Ewell, Marcel Goldschen-Ohm and Istvan Mody
for their assistance and suggestions on this project, along with appreciation to
Ejear Editing services.
Funding
This work was supported by grants from the Epilepsy Foundation (KPM, MVJ) and
NIH (NS046378, NS075366 to MVJ).
Conflict of Interest
The authors disclose that KPM, MVJ, ABN, and CC are included on the United States
patent US9629853B2 titled “Uses of Ganaxolone”. The authors declare no conflict of interest.
