
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy is a rare form of inherited cerebral small vessel disease associated with mutations in the high-temperature requirement serine peptidase A1 gene. As of now, only about 50 cases have been reported. In 2012, our group reported a family with a novel mutant of the high-temperature requirement serine peptidase A1 gene in China for the first time. To further explore the molecular pathogenesis of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, a recombination mouse model expressed human high-temperature requirement serine peptidase A1 gene mutant identified by our group was generated using the Donor & Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 system and termed the Mut-high-temperature requirement serine peptidase A1 geneL364P mouse model. Results show that Mut-high-temperature requirement serine peptidase A1 geneL364P mice present similar pathological characteristics to patients with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, suggesting that the Mut-high-temperature requirement serine peptidase A1 geneL364P mouse model was generated successfully. Moreover, apoptosis was induced in mouse brain vascular smooth muscle cells derived from Mut-high-temperature requirement serine peptidase A1 geneL364P mice. In summary, the cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy mouse model described in this study will be beneficial to demonstrate the pathological mechanism of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy and provide new therapeutic targets for clinical treatment.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is a rare inherited vascular disease (Nozaki et al., 2014). Patients with CARASIL show young-adult-onset non-hypertensive stroke, progressive motor and cognitive impairment, alopecia and lumbago (Fukutake, 2011; Fukutake and Hirayama, 1995; Nozaki et al., 2014). Brain Magnetic Resonance Imaging (MRI) has demonstrated diffuse leukoencephalopathy and multiple subcortical infarcts (Fukutake, 2011; Nozaki et al., 2014, 2015). Moreover, the autopsy findings of CARASIL have showed concentric thickening of the vascular wall, narrowing of the lumen, mild fibrous proliferation of the intima, and extensive loss of smooth muscle cells in the brain, which was similar with arteriosclerosis (Arima et al., 2003; Nozaki et al., 2014; Yanagawa et al., 2002). About 50% of patients were born to the consanguineous parents (Fukutake, 2011). Unfortunately, most of these patients die within 10 years of onset (Fukutake, 2011; Fukutake and Hirayama, 1995).
The first case of CARASIL was reported by a Japanese group in 1960 (Fukutake, 2011). Up until now, only about 50 cases have been reported and most of them were found in Japan (Fukutake, 2011; Nozaki et al., 2014). Additionally, 3 Indian families (Preethish-Kumar et al., 2017), 3 Chinese families (Chen et al., 2013; Wang et al., 2012; Xie and Zhang, 2018), 3 Caucasian families (Bianchi et al., 2014; Favaretto et al., 2019; , Mendioroz et al., 2010) and 1 Turkish family (Bayrakli et al., 2014) with CARASIL have been identified. In 2009, Japanese researchers revealed that CARASIL was associated with mutations in the high-temperature requirement serine peptidase A1 (HTRA1) gene on chromosome 10q through genome-wide linkage analysis (Hara et al., 2009). There are 9 exons in HTRA1 gene. To date, 6 missense mutations and 4 nonsense mutations have been reported and they are located in exon 1, 3, 4 and 6 of HTRA1 (Bayrakli et al., 2014; Bianchi et al., 2014; Chen et al., 2013; Hara et al., 2009; Mendioroz et al., 2010; Nishimoto et al., 2011; Preethish-Kumar et al., 2017; Wang et al., 2012).
HTRA1 is a serine protease and represses signaling through transforming growth factor (TGF)-β family members, which are important for vascular integrity (Oka et al., 2004). Mutations of the HTRA1 gene lead to low levels of protease activity and the loss of HTRA1 protein (Hara et al., 2009). Loss of the HTRA1 activity or product results in an increase of TGF-β signaling in cerebral small arteries and the subsequent accumulation of TGF-β members and TGF-β signaling-induced protein, including an extra domain A fibronectin, versican, and hyaluronan (Fukutake, 2011; Hara et al., 2009; Nozaki et al., 2014; Oka et al., 2004). In addition, acceleration of TGF-β signaling might induce extensive loss of vascular smooth muscle cells observed in patients with CARASIL (Nozaki et al., 2014). Thus, it is considered that increased TGF-β signaling contributes to the pathogenesis of CARASIL (Fukutake, 2011). However, the underlying mechanism of TGF-β signaling causing CARASIL is still unclear.
A previous study in our group reported a CARASIL family and a novel HTRA1 mutant (c.1091 T > C, L364P) in China for the first time (Wang et al., 2012). Thus, we intended to explore the molecular pathogenesis of CARASIL. Here, a recombination mouse model expressed mutant human HTRA1 identified by our group is built by the Donor & Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system, which is helpful to uncover the pathological mechanism of CARASIL.
All animal experiments were approved by the Institutional Animal Care and Use Committee and the Animal Experimental Ethics Committee of the Shandong Normal University. C57BL/6J and CD1 mice were purchased from Model Animal Research Center of Nanjing University (Nanjing, China). All mice were acclimatized for a week without any experimentation. Mice had ad libitum access to water and food. Animals were maintained in a standard light-dark cycle with an ambient temperature of 22 ± 4 °C. Contributions.
After submitting the sequence containing the targeting region of mouse HTRA1 to CRISPR Design Tool (http://crispr.mit.edu/, Zhang Feng Lab), the sgRNA with the highest score was chosen and responding oligos were synthesized. Next, diluted oligos were used to ligate with linearized pX330 vector to construct expressing vector as previously described (Wang et al., 2018), and sgRNA was produced using the HiScribe™ T7 High Yield RNA Synthesis Kit (E2040S, NEB) according to the manufacturer’s protocols in vitro. The sequence of sgRNA is: 5`GCCGTCGGGGACCGGCCGCT3`.OCT Cardiovascular Image Preprocessing.
Template DNA was extracted from the hair of CARASIL patients identified by our group. Then the human HTRA1 serine protease domain with L364P mutant was amplified from template DNA using polymerase chain reaction (PCR) and confirmed by sequencing. Primers used are hHTRA1 F: 5` GAATTCCAGCTGTCCCGGGCCG3` and hHTRA1 R: 5`AAGCTTCTATGGGTCAATTTCTT3`. Next, the PCR product was cloned into the pGEM-3Z vector to generate the donor vector.
Collection of zygotes and microinjection of plasmids were performed as previously described (Zhang et al., 2015). sgRNA, Cas9 and donor vector for Human HTRA1 serine protease domain with L364P mutant recombination were diluted by injection buffer (each 5 ng/μl) and co-injected pronuclear. Zygotes were then transferred to the pseudo-pregnant CD1 females at two-cell stage.
The genotyping of mice was performed by tail PCR and sequencing. Tail clippings were collected and digested in GNT-K buffer at 55 °C overnight. Tail preps were then diluted and boiled for 15 min. To fully screen founder mice, primers for WT (Fig. 1B, #1), inserted fragment (Fig. 1B, #2), 5` arm of target region plus partial inserted fragment (Fig. 1B, #3) and 3` arm of target region plus partial inserted fragment (Fig. 1B, #4) were used. Primers used for PCR were listed in Table 1.
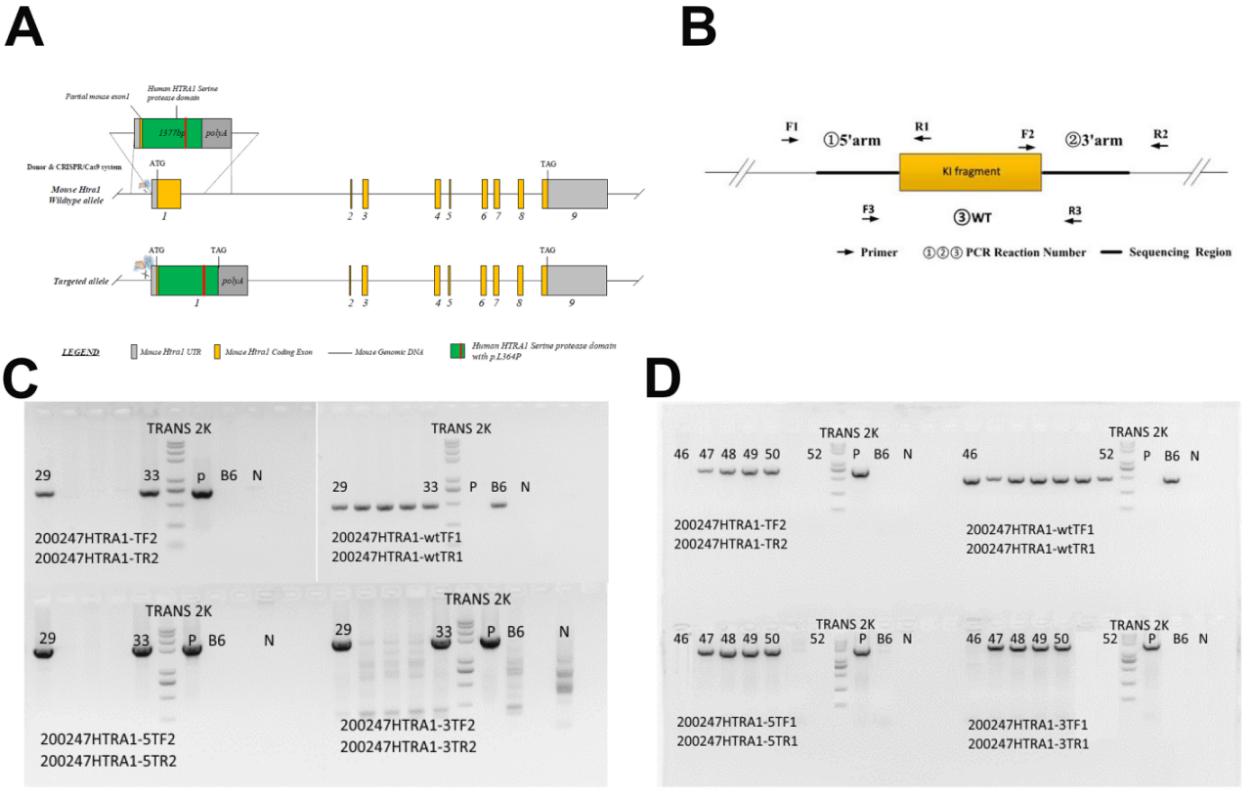 Figure 1.
Figure 1.Generation of heterologous recombination mice.
(A) Targeting strategy of HTRA1 gene. (B) Designed primer pairs for screening recombination mice. #1 was designed for WT region, #2 was designed for inserted fragment, #3 was designed for 5` arm of target region plus partial inserted fragment and #4 was designed for 3` arm of target region plus partial inserted fragment. (C) Recombination screening by PCR.B6: negative control of which template is genomic DNA of C57BL/6J mice; N: blank control without template; TRANS 2K PLUS II marker size: 8000bp, 5000bp, 3000bp, 2000bp, 1000bp, 750bp, 500bp, 250bp, 100bp.
| Primer Name | Sequence (5`-3`) | GC% | Tm | Expected Band Size | Illustration |
|---|---|---|---|---|---|
| 200247HTRA1-wtTF1 | AGAGAAGCACTGCGGGGTGT | 60 | 56 | KI/kI = 0 bp; |
#1 |
| 200247HTRA1-wtTR1 | GAGGAAGAGGGCCGGAGTTAC | 61.9 | 55.7 | ||
| 200247HTRA1-TF2 | ACCCTGAACATGGTGGTCCG | 60 | 56.7 | KI/kI = 720 bp; |
#2 |
| 200247HTRA1-TR2 | ACCAGGCACACATGTCAGACG | 57.1 | 55.6 | ||
| 200247HTRA1-5TF1 | GCCAGGCACAGAGATCTGCC | 65 | 57 | KI = 2070 bp; |
#3 |
| 200247HTRA1-5TR1 | GCTGGACGTGAGTGACATCAT | 65 | 58.3 | ||
| 200247HTRA1-5TF2 | CTCAGGTGGGGACAGCACATAC | 59 | 55.8 | KI = 2196 bp; |
#3 |
| 200247HTRA1-5TR2 | ACGCTCCTGAGATCACGTCTG | 57.1 | 54.2 | ||
| 200247HTRA1-3TF1 | ACTCGGGAGGCCCGTTAGTA | 60 | 55.1 | KI=2010bp; |
#4 |
| 200247HTRA1-3TR1 | GCTGTGTGATGGTCAGCTGC | 60 | 53.8 |
Cerebral arteries were collected from WT and recombination mice and then digested. Primary culture of mouse brain VSMCs was performed as previously described (Chi et al., 2017).
VSMCs and cerebral arteries collected from WT and recombination mice were fixed in 4% paraformaldehyde overnight. Next VSMCs were antigen-unmasked using citrate buffer and blocked in 5% bovine serum albumin in PBST (0.1% Triton X-100) for 1 hour. The following antibodies were used as primary antibodies: SM22α (1 : 200) (Cell Signaling Technology, Boston, USA) and human HTRA1 (1 : 200) (Abcam, New Jersey, USA). Normal rabbit IgG was used at the same dilution. Finally, cells were counterstained with DAPI. Images were captured using an inverted microscope (Nikon Eclipse E1000M).
To evaluate the status of cerebral stroke in recombination mice, neurological test was performed as described previously (Zhang et al., 2002). Briefly, a four point neurological score was used to evaluate the status of mice: 0 = normal, 1 = failure to extend the left forepaw fully, 3 = circling to the left, 4 = unable to walk spontaneously.
Cerebral arteries from WT and recombination mice were collected and fixed in methacarn fixative solution and then embedded in paraffin. The paraffin sections were then cut into 5 μm sections. After deparaffinization and rehydration, all sections were underwent hematoxylin and eosin (H & E) staining through standard protocol. Sections were then photographed and examined.
Using the Trizol reagent (Invitrogen), mRNA was isolated from brain VSMCs and reversed transcribed by the QuantiTect Reverse Transcription Kit for mRNA (Qiagen, Germany). Real-Time PCR was performed using the Maxima SYBR Green/ROX qPCR Master Mix assay (Thermo Scientific, Shanghai, China) in the StepOnePlus system (Applied Biosystem). Primer sequences are listed as follows: RT-hHTRA1 F: 5`CAGCTGTCCCGGGCCG3` and RT-hHTRA1 R: 5`CTATGGGTCAATTTCTT 3`.
Briefly, protein extracted from cerebral VSMCs were resolved by SDS-PAGE equally and subsequently transferred onto the Immobilon-P membranes (Millipore, Shanghai, China). 2 mg Precision Plus Protein Standards obtained from Bio-Rad Laboratories (Hercules, CA, USA) were loaded into the first lane of the gel. After transmembrane, the membranes were incubated in 5% nonfat milk for 1h at RT and then at 4 °C with primary antibodies in 5% nonfat milk overnight. Following primary antibody incubation, the membranes were exposed to secondary antibodies. The signals of interested protein were then detected by SuperSignal West Femto Maximum Sensitivity Substrate kit (Thermo Scientific, Shanghai, China) according to manufacturer’s instructions. HTAR1 primary antibody was purchased from Abcam (ab199529).
Brain VSMCs apoptosis was detected by Roche TUNEL Kit (Mannhein, Germany) following the manufacturer’s protocol.
2×106 brain VSMCs from each group were collected and washed twice by incubation buffer containing 10 mmol/L HEPES/NaOH, pH 7.4, 140 mmol/L NaCl, 5 mmol/L CaCl2. Next, cells were resuspended into 100 μl PBS supplementary with 1.5 μg/ml Annexin V (Becton Dickinson, New Jersey, USA) and moderate Propidium iodide (PI) (Thermo Scientific), and then incubated at room temperature for 10-15 minutes avoiding light. After washing, the cells were resuspended by incubation buffer. Apoptosis was analyzed by flow cytometry.
MTT assay kit (Abcam, Cambridge, UK) was used to detect brain VSMCs activity. Medium was discarded from cell culture in 96 well plates. Then 50 µL of serum-free media and 50 µL of MTT solution were added into each well and incubated at 37 °C for 3 hours. After incubation, 150 µL of MTT solvent was added into each well. Next the plate was shaken on an orbital shaker for 15 minutes and absorbance was measured at OD = 590 nm within 1 hour.
All values were presented as the mean SEM of individual samples. In the ANOVA analysis performed by the SigmaStat 3.5 software, a Tukey’s multiple-comparison test was adopted to estimate the significance of all data. Statistical significance was accepted when P < 0.05.
We co-injected the targeting plasmid encoding sgRNA for mouse HTRA1 serine protease domain and the donor vector with human HTRA1 serine protease domain containing L364P mutant into mouse zygotes to generate the recombination mice (Fig.1A). A total of 50 zygotes were injected and cultured. 34 zygotes developed to the two-cell stage and were transfected to 3 pseudo-pregnant CD1 mice. Among the 30 full term pups, 2 contained targeted recombination detected by four pairs of primers (Fig. 1B). These two founder female mice (F0 generation) were backcrossed with the C57BL/6J strain to generate the heterologous recombination mice (F1 generation). In total four heterologous recombination mice termed Mut-HTRA1L364P mice were generated (Fig. 1C, Table 2). Subsequently, more heterologous recombination mice were generated for the following experiments.
| ID | Gender | Color | Genotype | Generation |
|---|---|---|---|---|
| 29 | ♂ | Black | KI/wt | F1 |
| 33 | ♀ | Black | KI/wt | F1 |
| 47 | ♂ | Black | KI/wt | F1 |
| 48 | ♂ | Black | KI/wt | F1 |
CARASIL is a rare vascular disease, thus we detected the expression of human HTRA1 in mouse brain VSMCs. Primary cultured VSMCs were used in this study. To identify the brain VSMCs obtained by the primary culture, the marker smooth muscle SM22alpha (SM22α) was used for immumostaining. Results show that the cultured cells were VSMCs expressing SM22α (Fig.2A). Next, the human HTRA1 mRNA in VSMCs was detected by specific primers. Compared to the VSMCs from Mut-HTRA1L364P mice, no human HTRA1 mRNA was found in VSMCs of WT mice (Fig.2B). In addition, human HTRA1 protein was also not detected in VSMCs derived from WT mice while present in those of Mut-HTRA1L364P mice (Fig. 2C). Thus, human HTRA1 can be expressed in recombination mice.
 Figure 2.
Figure 2.Expression of human HTRA1 in brain VSMCs.
(A) Immunostaining of SM22α (green) in primary-cultured mice brain VSMCs. All cell nuclei were stained with DAPI (blue). (B) mRNA level of human HTRA1 in primary-cultured mouse brain VSMCs. (C) Protein abundance of human HTRA1 in primary-cultured mouse brain VSMCs. Bar graphs for protein abundance were quantitative data from three independent experiments. WT: wild-type mice; Mut-HTRA1L364P: recombination mice with human HTRA1 serine protease domain containing L364P mutant.*** indicates significant difference compared with the WT group (P < 0.05).
To further identify the location of human HTRA1, the immunofluorescence was performed using human HTRA1 antibody. Results showed that human HTRA1 was expressed in cytoplasm (Fig. 3), consistent with the location of human HTRA1 in human cells. This data suggested that the Mut-HTRA1L364P mice could be helpful to explore the pathological mechanism of CARASIL.
 Figure 3.
Figure 3.Location of human HTRA1 in mouse brain VSMCs. Immunostaining of human HTRA1 (red) in primary-cultured mouse brain VSMCs. All cell nuclei were stained with DAPI (blue).WT: wild-type mice; Mut-HTRA1L364P: recombination mice with human HTRA1 serine protease domain containing L364P mutant.
Similar to patients with CARASIL, Mut-HTRA1L364P mice also presented stroke, epilation, and vascular abnormality (Table 3). Neurological deficit scores were employed to assess neurological outcome affected by stroke in mice. Mut-HTRA1L364P mice exhibited elevated neurological deficit scores compared to those obtained from WT mice (Fig. 4A), suggesting the high risk of stroke. Moreover, cerebral arteries of Mut-HTRA1L364P mice show concentric thickening of vascular wall and disorder structure, consistent with that observed in patients with CARASIL (Fig. 4B). All of this data demonstrated that Mut-HTRA1L364P mice had similar pathological characteristics to patients with CARASIL.
 Figure 4.
Figure 4.Pathogenesis of Mut-HTRA1L364P mice.
(A) Neurological deficit scores of mice. (B) H&E staining of paraffin-sectioned cerebral arteries collected from mice. WT: wild-type mice; Mut-HTRA1L364P: recombination mice with human HTRA1 serine protease domain containing L364P mutant. *** indicates significant difference compared with the WT group (P < 0.05).
| Number | Gender | Stroke | Epilation | Vascular abnormality |
|---|---|---|---|---|
| 1 | male | N | N | N |
| 2 | male | N | N | N |
| 3 | female | Y | Y | Y |
| 4 | female | N | N | N |
| 5 | male | N | N | N |
| 6 | female | N | N | N |
| 7 | female | N | N | N |
| 8 | male | N | N | N |
| 9 | male | Y | N | Y |
| 10 | female | N | N | N |
| 11 | male | N | N | N |
| 12 | male | Y | Y | Y |
| 13 | male | N | N | N |
| 14 | female | N | N | N |
| 15 | female | N | N | N |
It is well known that CARASIL induces extensive loss of VSMCs in patients. However, the underlying mechanism is still unclear. Through MTT assay and FACS, we found that the activity and proliferation of brain VSMCs from Mut-HTRA1L364P mice were dramatically reduced (Fig. 5A and B). In contrast, VSMCs apoptosis was significantly induced in Mut-HTRA1L364P mice (Fig. 6A and B). Therefore, CARASIL may induce extensive loss of VSMCs through activating apoptosis.
 Figure 5.
Figure 5.Cell activity and proliferation of mouse brain VSMCs.
(A) Activity of mouse brain VSMCs detected by MTT assay. (B) Cell proliferation of mouse brain VSMCs detected by FACS. Bar graphs for cell proliferation were quantitative data from three independent experiments. WT: wild-type mice; Mut-HTRA1L364P: recombination mice with human HTRA1 serine protease domain containing L364P mutant. *** indicates significant difference compared with the WT group (P < 0.05).
 Figure 6.
Figure 6.Cell apoptosis of mouse brain VSMCs.
(A) FACS analysis of primary-cultured mouse brain VSMCs for apoptosis and quantification of apoptotic cells per group. Annexin V labeled with FITC and PI were used to stain cells. Bar graphs for cell apoptosis were quantitative data from three independent experiments. (B) Representative images showing apoptotic level in primary-cultured mouse brain VSMCs by TUNEL. Apoptotic cells were labeled in red and nuclei were labeled in blue by DAPI. WT: wild-type mice; Mut-HTRA1L364P: recombination mice with human HTRA1 serine protease domain containing L364P mutant. *** indicates significant difference compared with the WT group (P < 0.05).
CARASIL is a single-gene disorder caused by mutations in the HTRA1 gene and affects the cerebral small blood vessels. In 2012, our group reported a CARASIL family with a novel HTRA1 mutant (c.1091 T > C, L364P) in China for the first time (Wang et al., 2012). To further explore the pathological mechanism of CARASIL, we generated a recombination mouse model expressing mutant human HTRA1 and termed it the CARASIL mouse model. Results shown that the Mut-HTRA1L364P mice presented similar pathogenesis to patients with CARASIL, suggesting that the CARASIL mouse model was generated successfully and should be useful for the research of CARASIL.
In this study we showed that the L364P mutant of HTRA1 also induced CARASIL in mouse. However, 6 missense mutations and 4 nonsense mutations distributed in exon 1, 3, 4 and 6 of HTRA1 respectively have been reported (Bayrakli et al., 2014; Bianchi et al., 2014; Chen et al., 2013; Hara et al., 2009; Mendioroz et al., 2010; Nishimoto et al., 2011; Preethish-Kumar et al., 2017; Wang et al., 2012). Therefore, the CARASIL mouse model generated by us may be not suitable for revealing the underlying mechanism how other HTRA1 mutations cause CARASIL. In future, we would generate more novel CARASIL mouse models with other HTRA1 mutations to uncover the causing mechanism of CARASIL completely.
Numerous studies have indicated that HTRA1 represses signaling through TGF-β family members, which play a vital role in vascular integrity (Favaretto et al., 2019; Shiga et al., 2011; Xie and Zhang, 2018). To further identify the characteristics of the CARASIL mouse model, we need to detect the level of TGF-β members and TGF-β signaling-induced proteins in the brain VSMCs. Moreover, the relationship between accumulation of TGF-β signaling in vessels and the pathology of CARASIL should be of focused in future studies.
TGF-β family members are closely associated with vessel morphogenesis and stability and play multiple roles in VSMCs (Grainger, 2004; ten Dijke and Arthur, 2007). Loss of HTRA1 activity or product leads to the accumulation of TGF-β members and TGF-β signaling-induced proteins in cerebral small arteries (Nozaki et al., 2014). Enhanced TGF-β signaling contributes to numerous vascular diseases, such as Marfan’s syndrome and hypertension (August and Suthanthiran, 2006; Loeys et al., 2005; Pannu et al., 2005). Therefore, it is suggested that accumulating TGF-β signaling may lead to vascular disorder in CARASIL.
Growing studies have identified that TGF-β members including TGF-β1 induce cell apoptosis through multiple pathways (Ferrari et al., 2006; Wassmer et al., 2006; Yan et al., 2018). Meanwhile, increased brain VSMCs apoptosis was observed in the Mut-HTRA1L364P mouse which may contribute to extensive loss of VSMCs induced by CARASIL. All these results together suggest that CARASIL may induce extensive loss of VSMCs through activating apoptosis by the accumulation of TGF-β signaling in cerebral small arteries.
TGF-β signaling-induced proteins are also enriched in cerebral small arteries of patients with CARASIL. However, these proteins including an extra domain A fibronectin, versican and hyaluronan protect vessel against dysregulation and apoptosis (Huang et al., 2006; Kenagy et al., 2006; Lisignoli et al., 2001; Ou et al., 2012; Sattar et al., 1994; Wu et al., 2005). In other words, TGF-β signaling-induced proteins play opposite roles in vessel compared to TGF-β family members. Thus, it is suggested that elevated TGF-β signaling due to HTRA1 mutations may suppress functions of TGF-β signaling-induced proteins on alleviating CARASIL-induced vascular disorder. These TGF-β signaling-induced proteins may be involved in other pathogenesis of CARASIL.
In summary, we generated a recombination mouse model expressed the L364P mutant of human HTRA1 gene and termed it the CARASIL mouse model. This CARASIL mouse model should be helpful to demonstrate the pathological mechanism of CARASIL and provide new therapeutic targets for clinical treatment.
This work was supported by Science and Technological Project of Shandong (Major innovation project. Grant No. 2018CXGC1502).
The authors declare that they have no competing interests, and all authors should confirm its accuracy.