

1 Shanghai Jiaotong University, 200030 Shanghai, China
2 Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 200030 Shanghai, China
3 Shandong University of Traditional Chinese Medicine, 250355 Jinan, Shandong, China
†These authors contributed equally.
Abstract
Frequent premature ventricular contractions (PVCs) can cause a reversible form of cardiomyopathy in patients without structural heart disease. Because of the challenging nature of PVC-induced cardiomyopathy (PVICM), the mechanisms and risk factors for PVICM are still unclear. Based on the evidence from retrospective and observational studies, the risk factors for the development of PVICM, in addition to PVC exposure, include QRS duration, coupling interval and male sex. Based on animal models, abnormal calcium handling and cardiac remodeling may be the crucial mechanism underlying the development of cardiomyopathy. We have summarized the current knowledge on PVICM in this review. Understanding these mechanisms and risk factors is important for the diagnosis and management of this condition, which can lead to heart failure if left untreated.
Keywords
- premature ventricular contraction
- cardiomyopathy
- mechanism
- risk factor
Premature ventricular contractions (PVCs) are implicated in the reversible cardiac heart failure referred to as PVC-induced cardiomyopathy (PVICM). Current guidelines and expert consensus suggest that a PVC load of 10–15% predisposes to PVICM due to impaired left ventricular function [1, 2]. There is growing clinical evidence that a PVC loading above 0.12% increases the risk of death by 31% [3]. Therefore, PVICM has become an important clinical issue that requires urgent attention. However, the exact pathophysiological mechanism of PVICM has not yet been fully clarified. Therefore, it is necessary to review the current mechanisms and risk factors associated with PVICM.
PVCs, defined as early depolarization of the myocardium originating in the
ventricle, are due to increased automaticity, triggered activity, or reentry.
According to large cohort studies, the prevalence of PVCs ranges from 1% to 4%
[4, 5], as derived from screening standard 12-lead electrocardiograms. The
diagnostic capability of a 12-lead electrocardiogram (ECG) has acknowledged limits. Based on a
2-minute ECG, PVCs are present in
PVCs are generally considered to be benign [7], despite the reciprocal relationship between arrhythmias and cardiomyopathy. In 1988, Duffee et al. [8] proposed that suppression of PVCs could improve left ventricular function in patients with presumed idiopathic dilated cardiomyopathy. Since then, several studies have reported the relationship between reversible left ventricular dysfunction and frequent PVCs. The first case of radiofrequency ablation in PVICMs was reported by Chugh et al. [9] in 2000, who noted resolution of the dilated cardiomyopathy after eliminating PVCs by ablation. There has been increased attention to the reversible cardiomyopathy caused by PVCs.
Frequent PVCs (
Clinically, patients with PVICM usually have elevated brain natriuretic peptide (BNP) in contrast to patients with only PVCs. Echocardiographic measurements in PVICM patients showed LV end-systolic wall thickening, increased inner diameters during systole and diastole, suggesting systolic dysfunction and LV remodeling. Some changes, such as left ventricular dysfunction, mild cardiac fibrosis and electrical remodeling, were present in swine and canine models of PVICM, similar to those found in PVICM patients. In a PVICM canine model, eccentric hypertrophy was the typical cardiac remodeling after 12 weeks of pacing [17]. In terms of the cardiomyocyte morphology, the size of cardiomyocytes was larger in a PVICM swine model. The morphology of sarcomere, Z-line arrangement was disarrayed and the shear angles at the Z-line were reduced in the PVICM cardiomyocytes [18]. Compared to animal models, PVICM in humans usually develops over several years [16] (Table 1, Ref. [18, 19, 20, 21, 22, 23, 24, 25]).
| Structural changes | |||||
| LVEF | Decreased | ||||
| LV mass | Increased (LV end-systolic wall thickening) | ||||
| Larger size of CMs [18] | |||||
| fibrosis | mild | ||||
| Hemodynamic change [19, 20] | |||||
| stroke volume | Decreased during PVC | ||||
| overall cardiac output | |||||
| Autonomic dysregulation | extracardiac sympathetic hyperinnervation and sympathetic neural hyperactivity | ||||
| increased coronary sinus norepinephrine levels | |||||
| Myocyte remodeling | |||||
| T-tubules | Decreased [21] | ||||
| Z-line | Disarrayed, shear angles of z-line reduced | ||||
| dyad | Dyadic density decreased, JPH2 and BIN1 declined | ||||
| Conformation of myosin heads | unknown | ||||
| Electrical changes | |||||
| ICaL | |||||
| Ito | |||||
| IK1 | |||||
| Prolonged APD and exaggerated variations [21] | |||||
| Calcium signaling alteration | |||||
| Protein levels | Distribution | ||||
| Cav1.2 | Decreased | LV free wall; LV | |||
| SERCA2a | Decreased | LV free wall; LV | |||
| PLN | Increased | LV free wall; LV | |||
| pPLN | LV | ||||
| RyR2 | Increased | LV | |||
| NCX | Increased | LV: shift from dyads to peripheral sarcolemma [25] | |||
| CaMKII- |
Increased | basal-lateral LV | |||
| Dayd remodeling | |||||
| JPH2 | LV: dim and dispersed | ||||
| BIN1 | LV: dim and dispersed | ||||
The detection of cardiac hemodynamic indicators in different pacing modes found that cardiac output was significantly reduced in ventricular demand pacing or inhibited ventricular pacing (VVI) and dual-chamber demand pacing with dual-rate responsiveness (DDDR) modes compared with the atrial demand pacing or inhibited atrial pacing (AAI) mode [19]. pulmonary capillary wedge pressure (PCWP), right atrium (RA) pressure and pulmonary artery pressure all increased, and left ventricular output index was lower in the VVI mode [19]. LVEF was significantly decreased as assessed by radionuclide angiography [19]. There are similar changes in hemodynamics in patients with PVCs. The stroke volume decreased during the premature beat [20]. Although the post-systolic enhancement effect of the next heart beat might compensate for the lost output, the overall cardiac output is lower than that of the sinus beat. With the prolonged RR interval, the left ventricular end-systolic pressure caused by post-extrasystolic potentiation (PESP) increased significantly [26].
Over 50% of PVICM patients achieved improved cardiac function and symptom remission after eliminating PVCs with antiarrhythmic drugs and ablation [12]. Treatment in PVC patients can improve LV diastolic function and left atrial function in the short term. Animal experiments found that myocardial interstitial fibrosis, autonomic dysregulation and LV mechanical dyssynchrony persist for a few weeks after eliminating PVCs, despite improvement in LV function [25]. Further studies found electrical remodeling in PVICM during sinus rhythm persists [27]. These irreversible changes might account for the higher risk of sudden death, malignant arrhythmias, and heart failure in patients who have had PVICM or PVCs [28].
The underlying mechanism of PVICM remains controversial. PVICM was initially classified as a tachycardia-induced cardiomyopathy because of the reversible cardiac function associated with arrhythmias [9]. Nevertheless, heart rate did not increase substantially in patients with PVCs according to ECG and 24 h-Holter monitoring, and except for the interval PVC that can increase heart rate, most systolic PVCs are invalid heartbeats. In comparison to cardiac remodeling in animal models of atrial and ventricular contractions, it was determined that the development of PVICM was not related to tachycardia and heart rate irregularity. Previous studies have consistently suggested that probable mechanisms involved in PVICM include abnormal calcium handling, dyssynchronous ventricular contraction, autonomic dysregulation and myocardial remodeling (Table 1).
Excitation-contraction coupling is a process providing the basis for muscle
contraction, in which the key effector molecule is calcium ions. Calcium-induced
calcium release (CICR) is a fundamental cellular mechanism for generating and
amplifying intracellular calcium signals. The excitation-contraction coupling of
cardiomyocytes depends on this process. PVCs lead to prolonged action potential
time of ventricular cardiomyocytes and a decrease in the density of the transient
outward K
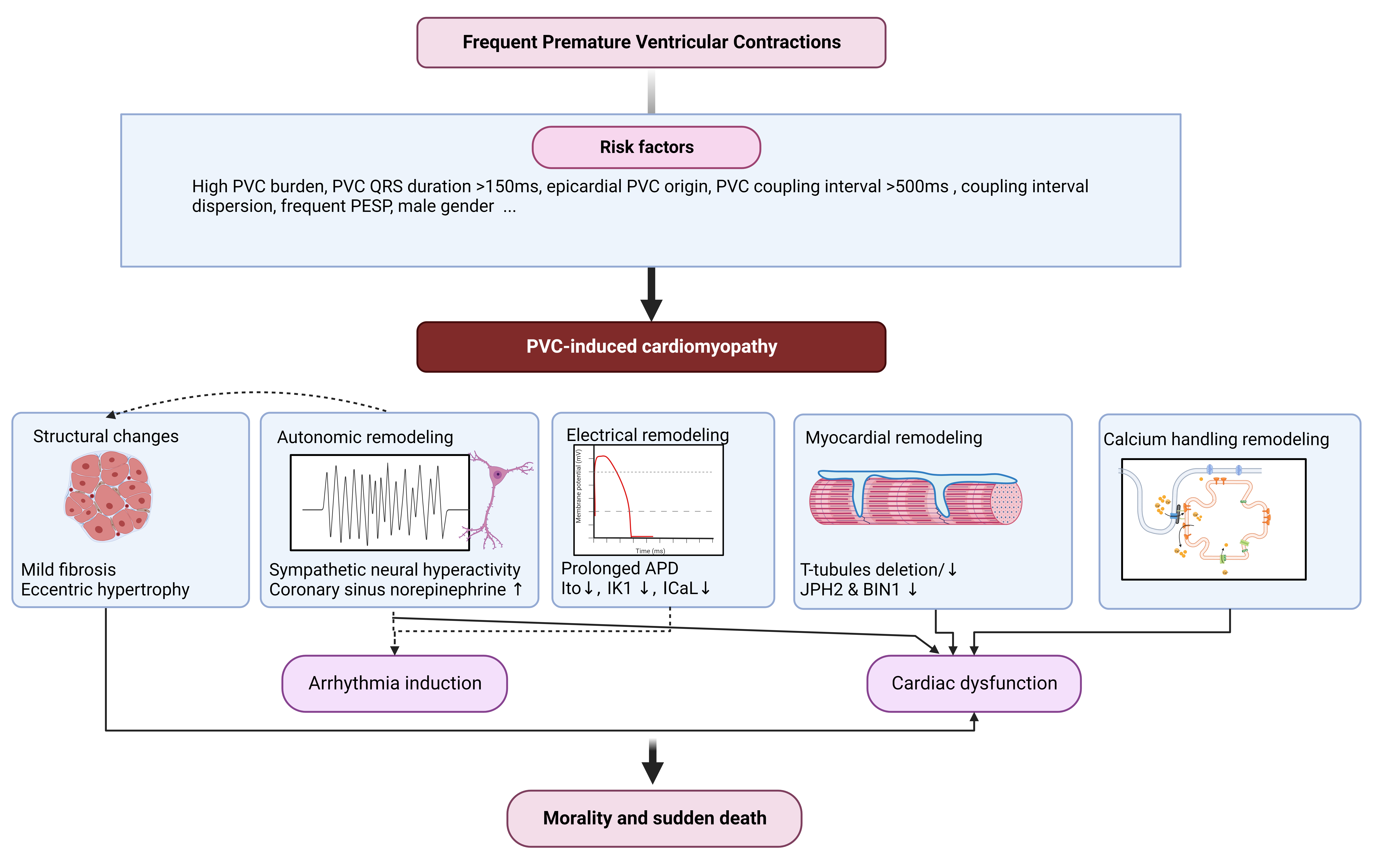
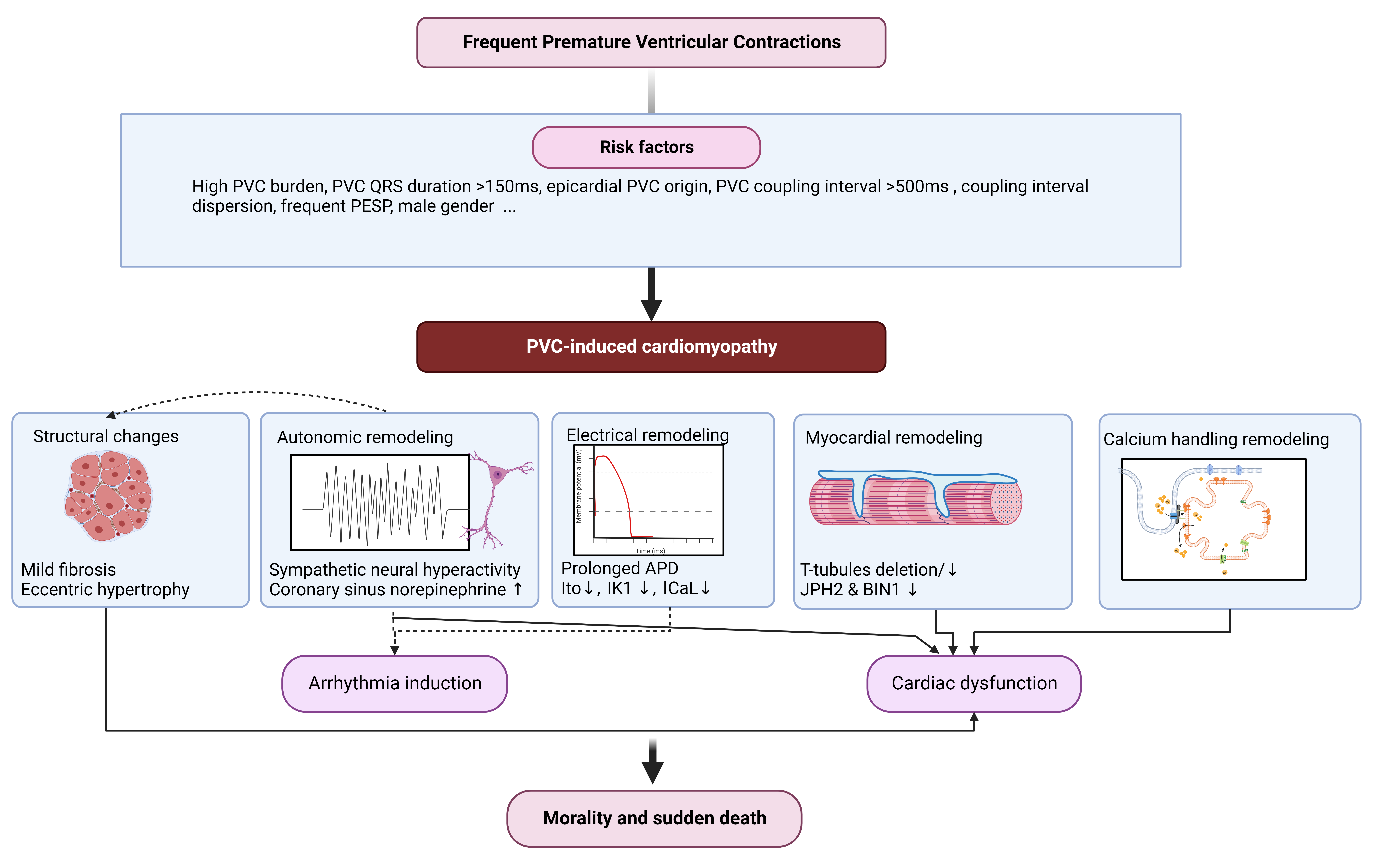 Fig. 1.
Fig. 1.premature ventricular contractions (PVCs) is the most common cardiac arrhythmia in patients, while
some of them could develop cardiac dysfunction in the several years. According
to different characteristics in patients, risk factors for premature ventricular contraction-induced cardiomyopathy (PVICM) are complex and
diverse, including PVC burden, PVC QRS duration PVC origin and sexuality.
Investigation of molecular mechanisms has predominantly been studied in animal
models, primarily swine and canine. Based on those models, typical tissue
alterations in PVICM are mild fibrosis and eccentric hypertrophy. Besides,
frequent PVCs enhance sympathetic activity, further exacerbating structural
alteration in models. Substructural remodeling includes reduced T-tubules,
decreased dyad intensity and Z-line arrangement disarray, leading to reduced
L-type calcium currents and decreased systolic calcium transient synchrony. All
of the changes might function corporately or separately, resulting in cardiac
dysfunction and malignant arrhythmia, increase the risk of sudden cardiac death.
PESP, post-extrasystolic potentiation; JPH2, junctophilin-2; BIN1, bridging integrator-1;
APD, action potential duration; Ito, transient outward K
Genome-Wide Association Studies (GWAS) confirm the association of genes coding for calcium handling proteins are at an increased risk of PVICM development in patients with frequent PVCs. It is hypothesized that mutations in calcium handling genes may affect calcium homeostasis, resulting in decreased sodium current and slow conduction, thereby prolonging the QRS duration [33].
Dyssynchronous ventricular contractions have long been considered to be the
primary mechanism for the development of PVICM. In PVICM animal models, a linear
relationship was found between the degree of LV dyssynchrony and the upregulation
of CaMKII-
The 24-hour electrocardiogram analysis of heart rate variability in patients with idiopathic PVCs found that autonomic activities were involved in the occurrence of PVCs [34, 35], and frequent PVCs lead to increased peripheral tissue and cardiac sympathetic activity and increased coronary sinus norepinephrine levels [26]. In a swine model of PVICM, neural remodeling was characterized by extracardiac sympathetic hyperinnervation and sympathetic neural hyperactivity [36]. The neural remodeling—stellate ganglia hyperinnervation—persisted despite normalization of LV systolic function [37]. Cardiac dysfunction in PVCs may be triggered and facilitated by chronic disruption of the sympathetic-vagal balance. At an early stage, depletion of cardiac transient receptor potential vanilloid-1 (TRPV1) afferents by resiniferatoxin (RTX) improved LV systolic function and alleviated cardiac fibrosis in PVICM animals, while this improvement was not apparent at late stages, and had no effect on autonomic activity [38]. These studies suggest that TRPV1 mainly plays a role in promoting myocardial fibrosis in the early stage, and then myocardial remodeling and dysfunction are mainly affected by sympathetic imbalance, indicating that neuromodulation may play distinct roles at different stages of PVICM.
The sliding filament theory is a classic model for explaining muscle contraction proposed by Andrew Huxley and Hugh Huxley in 1954, and is almost universally accepted [39, 40]. The cross-bridge cycling results in force production through cyclical conformational changes of myosin heads. The structure of myosin-actin interaction in the cross-bridge cycling has different characteristics and different spatial positions from thin filaments, as determined by X-ray diffractometry and scanning electron microscopy. The state of myosin heads close to the thin filament is defined as the disordered relaxed state; the super relaxed state is a low-energy metabolic state, where myosin head interacts with one another and the blocked head (BH) interacts with the lever of thick filaments, keeping the myosin head away from the filament and making it difficult to bind adenosine triphosphate (ATP) [41, 42]. It is currently believed that myosin super relaxed state (SRX) plays an important role in regulating energy utilization and cardiac contraction [43]. The cellular basis for the Frank-Starling mechanism is length-dependent activation (LDA). Calcium sensitivity increases when sarcomeres are stretched, causing increases in cardiac contractility. The mechanism of myofilament LDA derived from swine ventricular myocytes is that passive stretching converts more myosin SRX to disordered relaxed state (DRX) [44]. Changes in the balance between SRX/DRX may explain cardiac dysfunction occurring in cardiac diseases. Studies on familial hypertrophic cardiomyopathy have found that most variants are located in genes encoding myosin head and neck of filaments [45, 46, 47]. The structural changes of thick filaments change the DRX/SRX balance, and more myosin heads are in the DRX state resulting in cardiac hypercontractility and impaired diastolic function. Furthermore, Mavacamten, a myosin inhibitor that favors the closed conformation of myosin heads, achieved significant therapeutic effects in a Phase III clinical trial in obstructive hypertrophic cardiomyopathy [48]. In dilated cardiomyopathy, the aspartate-to-alanine substitution at position 94 in the regulatory light chain of myosin (RLC) encoded by the myosin regulatory light chain 2 (MYL2) gene results in an increased number of SRX heads and a subsequent reduction in myocardial contractility [45]. In addition to calcium handling, experimentally, myocardial force production is also modulated by alterations of conformations of the myosin head during ischemia, hypoxia or stretching. Therefore, it is important to explore the role of DRX/SRX in reversible cardiomyopathies such as PVICM and the efficacy of myosin inhibitors in PVICM.
Studies of myocardial cytoskeletal proteins in heart failure (hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM)) have suggested that cytoskeletal proteins, such as actin and desmin were significantly reduced [49, 50]. Similar changes in the actin cytoskeleton were observed in a swine model of PVICM, which was associated with Z-line arrangement disarray and played a crucial role in inefficient contractile function and cardiomyocyte remodeling [18]. This adaptive change may initially protect the heart from changes in mechanical stress to adapt to pressure changes, but long-term, there is a lack of adaptation and a decrease in the systolic and diastolic capacity of the heart. Similar alterations were observed in PVICM.
Dyads are subcellular structures of calcium signaling in cardiomyocytes, where the interconnection and connection of the plasma membrane network composed of the SR and the T tube, act as the intracellular calcium synapse of cardiomyocytes. In PVICM, the number of T-tubules is highly reduced, while the length of sarcomere and mitochondrial structure do not change significantly [22]. Decreased T-tubules diminish dihydropyridine receptor-ryanodine receptor (DHPR-RyR) coupling efficacy, which is responsible for reduced L-type calcium currents and decreased systolic calcium transient synchrony [22]. T-tubule remodeling occurs before the onset of ventricular dysfunction [51]. Previous studies suggest that the decreased expression of structural proteins such as junctophilin-2 (JPH2) and bridging integrator-1 (BIN1) is implicated in dyad formation [22, 52]. The N-terminal of JPH-2 is attached to the cell membrane through the MORN domain, and the C-terminal transmembrane structure anchors the SR, maintaining the stability of the SR and T tubular membrane structure. BIN1, as a membrane scaffold protein, plays an important role in the formation and structural maintenance of T-tubules. Decreased expression of both structural proteins will cause dyad abnormalities. It was hypothesized that increased stress on the local ventricular wall during PVCs was responsible for decreased expression of junctophilin-2 in cardiomyocytes and the remodeling of T-tubules [53] (Fig. 1).
Clinically, most frequent PVCs do not develop into a cardiomyopathy. Domestic and foreign guidelines have not clearly defined the need for eliminating PVCs to prevent cardiac dysfunction. It is still recommended that PVC patients with a high risk of PVICM undergo serial echocardiography to evaluate the changes in cardiac structure and function.
The risk factors for developing PVICM include PVC burden, QRS duration, PVC origin, interpolated PVCs, and male sex (Table 2, Ref. [15, 28, 54, 55, 56, 57, 58, 59, 60, 61, 62]). However, the effects of PVICM remain controversial.
| Del Carpio Munoz F et al. [54] | Ghannam et al. [55] | Kawamura et al. [56] | Bas et al. [57] | Yokokawa et al. [58] | Sadron Blaye-Felice et al. [59] | Voskoboinik et al. [60] | Olgun et al. [61] | Billet et al. [62] | Limpitikul et al. [15] | |
| Number of patients | 17 | 120 | 51 | 43 | 113 | 96 | 39 | 21 | 17 | 29 |
| PVC burden | 29.3% | 22% | 19% | 19% | 26% | 30% | NS | NS | ||
| PVC QRS duration | NS | NS | - | NS | NS | |||||
| Sinus QRS duration | - | - | - | - | - | Long sinus QRS duration* | - | - | - | - |
| PVC origin | RV PVCs | NS | NS | NS | Epicardial PVCs | Epicardial PVCs | NS | - | NS | - |
| Coupling interval | - | - | Longer CI | - | - | Long CI | - | - | NS | |
| CI-dispersion | - | - | 115 ms (maximum-CI–minimum-CI) | - | - | - | - | - | - | + |
| Interpolation | - | - | - | More frequent | - | + | - | + | NS | - |
| PESP | - | - | - | - | - | - | - | - | High PESP | - |
| Male sex | NS | + | NS | + | + | + | + | NS | NS | NS |
*In swine models of PVICM, sinus QRS duration increased significantly in the LV
Epi PVC (p
PVC burden has been the most consistent parameter to demonstrate a relationship
with the development of PVICM. Compared with PVC patients with normal ejection
fraction, PVICM patients tend to have higher PVC burden (16–30% per day)
[28, 54, 56, 61, 63, 64]. The lowest PVC burden resulting in cardiomyopathy was 10%
[65]. Baman et al. [65] found that a PVC burden of
High PVC burden could serve as a predictor for the development of PVICM.
However, it is still unclear why some patients do not develop cardiomyopathy
despite a high PVC burden and why some patients appear to develop PVICM with a
burden threshold of
The PVC QRS duration in PVICM patients was significantly longer than that in
patients without PVICM (164
Abnormalities of Calcium handling related proteins interrupt calcium homeostasis and are associated with prolonged QRS duration [33]. Therefore, it is thought that changes of QRS duration in PVCs suggest abnormal calcium handling in ventricular cardiomyocytes, ultimately resulting in PVICM.
The site of the PVC origin has an impact on the PVC QRS width and the degree of cardiac asynchrony. LV dysfunction was observed with PVCs from all common anatomic regions of origin. In contrast, PVCs originating from the epicardium appear to cause a more pronounced LV dyssynchrony and to induce LV systolic dysfunction, with longer QRS duration [58, 69]. PVCs originating from the RV were more likely to induce cardiac enlargement and compromise cardiac function [54]. However, results in other studies were contradictory, negating the association of PVC origin with the PVICM [66, 70]. It is commonly assumed that cardiac dyssynchrony is related to the PVC origin, but some studies have found that the degree of dyssynchrony is mainly dependent on the coupling interval rather than the origin [70].
In a previous study, a PVC coupling interval
The burden of interpolated PVCs was higher in the PVICM patients compared with other PVC patients. Interpolation of PVCs can independently predict PVICM-induced cardiomyopathy (odds ratio 4.43, 95% confidence interval 1.06–18.48, p = 0.04) [61].
PESP is defined as a physiological phenomenon of the increase in contractility following an extrasystole, and was first proposed by Oscar Langendoff in 1885. The cellular mechanism of PESP is that calcium release from intracellular stores is increased during the post-extrasystolic heartbeat. During the premature heartbeat, the transient decrease in calcium is caused by the refractoriness of RyRs. During the post-extrasystolic beat, RyRs have recovered from inactivation, and then increased intracellular calcium stores are released from these channels, resulting in increased contractility [73]. Previous studies found a significant increase in PESP in heart failure patients, and suggested that PESP could serve as a risk predictor of cardiac dysfunction and a prognostic indicator for patients with myocardial infraction [73, 74]. Increased PESP is associated with abnormal calcium cycling induced by heart failure. Similarly, patients with PVICM had a significantly higher PESP compared to controls [62]. In animal experiments, PESP was higher than at baseline after PVICM developed in canine models [75]. Furthermore, the level of PESP at baseline had a negative correlation with LVEF, suggesting that baseline PESP at the early stage of PVCs might be a predictor for PVICM [75].
In accordance with the multi-year follow-up results of multiple retrospective and prospective studies, males are at a greater risk of PVICM than females [76]. The reasons for the sex disparity in PVICM are still unclear. Female patients with frequent PVCs are more likely to experience symptoms such as palpitation and chest tightness than males, and they more often seek medical attention [77]. Furthermore, a longer history of palpitations and asymptomatic PVCs are independent risk factors for PVICM [59].
Speckle tracking image is a relatively non-invasive cardiac function imaging technology. Compared with conventional echocardiography, it can detect early myocardial structural changes before a decline in LVEF is detected by ECG. Global longitudinal strain (GLS) is a measure of LV global function that correlates with the extent of myocardial fibrosis. GLS appears to be useful to predict changes in LV function. Additionally, GLS is considered a prognostic indicator in PVICM, based on the association between mortality and GLS levels in cardiomyopathy patients [78].
Cardiac magnetic resonance imaging (CMR) is a noninvasive examination to assess
focal myocardial scar and diffuse myocardial changes. In PVC patients without
structural cardiomyopathy, the presence of myocardial scar was identified by
DE-CMR in 25% of patients with frequent PVCs, and it was independently
associated with the development of PVICM (odds ratio 2.2; 95% confidence
interval 1.3–3.7; p
There are several therapeutic interventions to prevent heart failure in PVC patients at high risk for PVICM. American guidelines recommend that catheter ablation is the treatment strategy for patients who experience symptoms and have decreased LV function due to frequent PVCs or who are unwilling to take antiarrhythmic drugs and for whom the antiarrhythmic drug (AAD) therapy is ineffective or the side effects of drugs are intolerable [1]. 2022 European Society of Cardiology (ESC) guidelines on ventricular arrhythmias (VA) and sudden cardiac death (SCD) recommend catheter ablation for first-line treatment for PVICM [80]. The results of clinical studies and animal experiments have confirmed that cardiac function was significantly improved when PVC burden was reduced. The degree of improvement is independent on the PVC origin. PVCs originating from the left and right hearts had similar benefits from successful rhythm control [81].
Catheter ablation therapy for PVICM patients has been reported to have an
immediate post-ablation success rate of 92.5% [82] and the long-term success
rate is 66%–90% [81, 83]. Catheter ablation has shown a high acute success
rate, and long-term monitoring has demonstrated significant reduction in PVC
burden, making it more effective than AADs [84, 85]. Furthermore, compared to
AADs, radiofrequency ablation can significantly improve left ventricular ejection
fraction (LVEF) in patients with PVCs (from 53% to 56%, p
There is still little effect for patients with myocardial scars identified by CMR before ablation [72]. Drug therapy should be considered for these patients who have poor postoperative outcomes or who do not receive catheter ablation, in order to reduce PVC burden if possible. Medical treatment to suppress the PVCs may include the use of beta-blockers or calcium channel blockers [80, 85] while the selection of antiarrhythmic drugs is limited by cardiomyopathy and heart failure. Attention should be paid to the long-term management of PVICM patients whose cardiac function is fully restored to normal. In congestive heart failure patients with ventricular arrhythmias, the application of amiodarone can effectively suppress ventricular arrhythmias and improve ventricular function. Unfortunately, it did not reduce the incidence of sudden death or improve survival [88]. “Dyssynchrony memory”, describing a phenomenon that LV dyssynchrony persists after PVC cessation, in the recovery period of PVICM swine, is a reminder for the need for long-term follow-up in patients with PVCs and PVIC to further our understanding of ventricular arrhythmias.
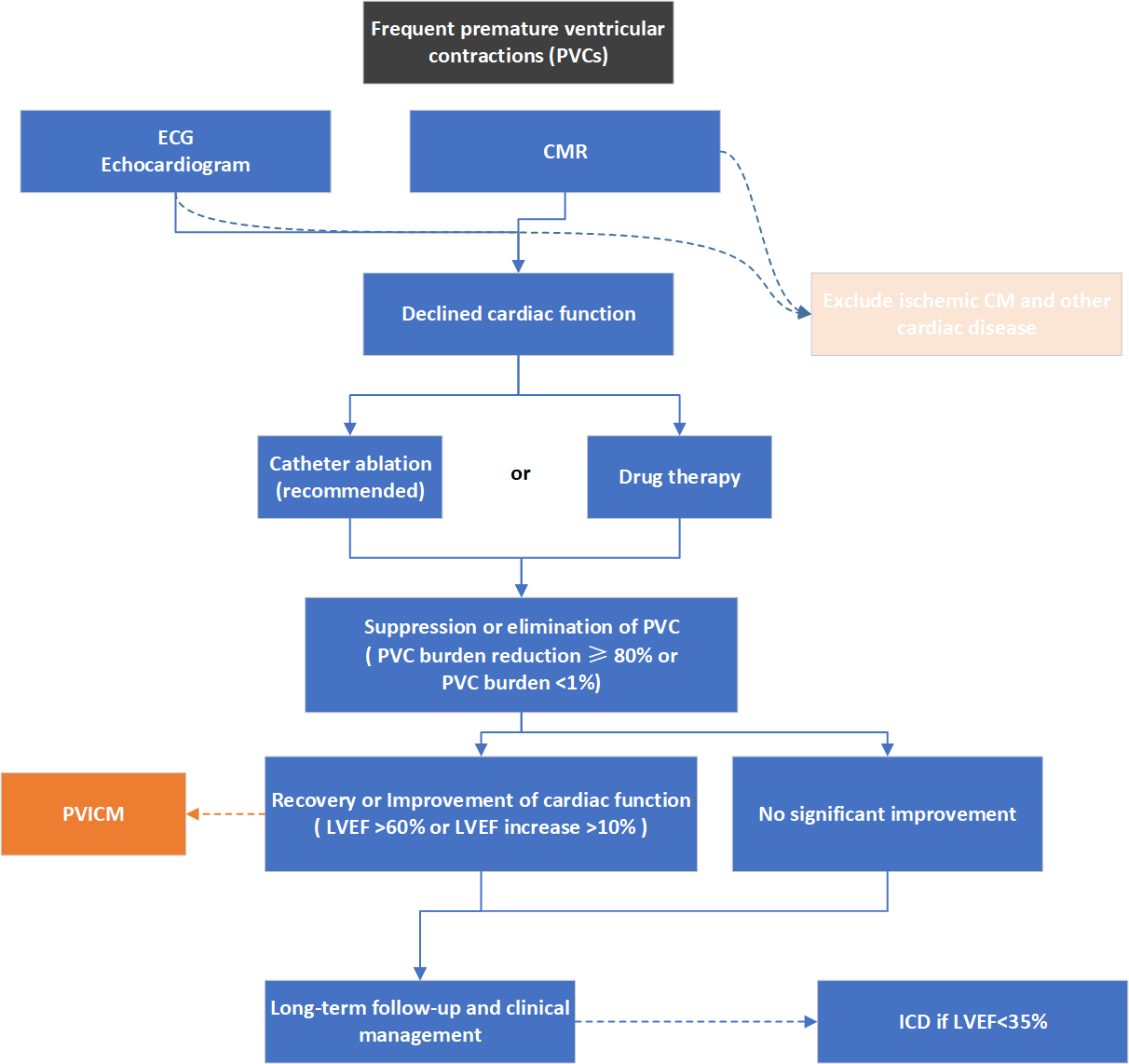
ECG and echocardiography are essential assessment tools in the long-term management of PVICM (Fig. 2). According to the time course of recovery of LVEF in PVICM patients, the greatest improvement was observed within one week after PVC ablation [12]. Yokokawa et al. [89] found that most patients (around 75%) who underwent successful ablation can recover LV function within 4 months, while a few may take several years (up to 45 months) for recovery. Moreover, multicenter studies on idiopathic PVC ablation showed that around 20% of patients may require repeat ablation, primarily for PVCs originating in epicardial and papillary muscle locations [86]. In 60 PVICM patients who underwent successful ablation of PVCs, 16.7% (10 patients) experienced recurrent PVCs, resulting in PVICM recurrence [90]. Based on these findings, short-term (within one month) evaluation of treatment efficacy and recovery of cardiac function should be performed after ablation or AADs therapy, and medication should be adjusted promptly for patients who have a poor response to therapy. PVCs may indicate worsening of underlying heart disease and exacerbate heart failure. PVICM patients often experience heart failure recurrence after PVC recurrence. Therefore, follow-up with ECG and echocardiography every three to six months should be conducted to assess cardiac function and adjust anti-arrhythmic and heart failure medications in a timely manner. In addition, PVCs may trigger malignant arrhythmias such as ventricular tachycardia (VT) in patients. A clinical study showed that among 30 PVICM patients, 9 cases (36%) had ECG findings suggestive of VT, including 3 cases of sustained ventricular tachycardia [12]. Thus, prevention of sudden cardiac death (SCD) events should be emphasized in PVICM patients, and implantable cardioverter-defibrillator (ICD) implantation should be considered based on patient symptoms, ECG changes, and compliance with treatment indications [80].
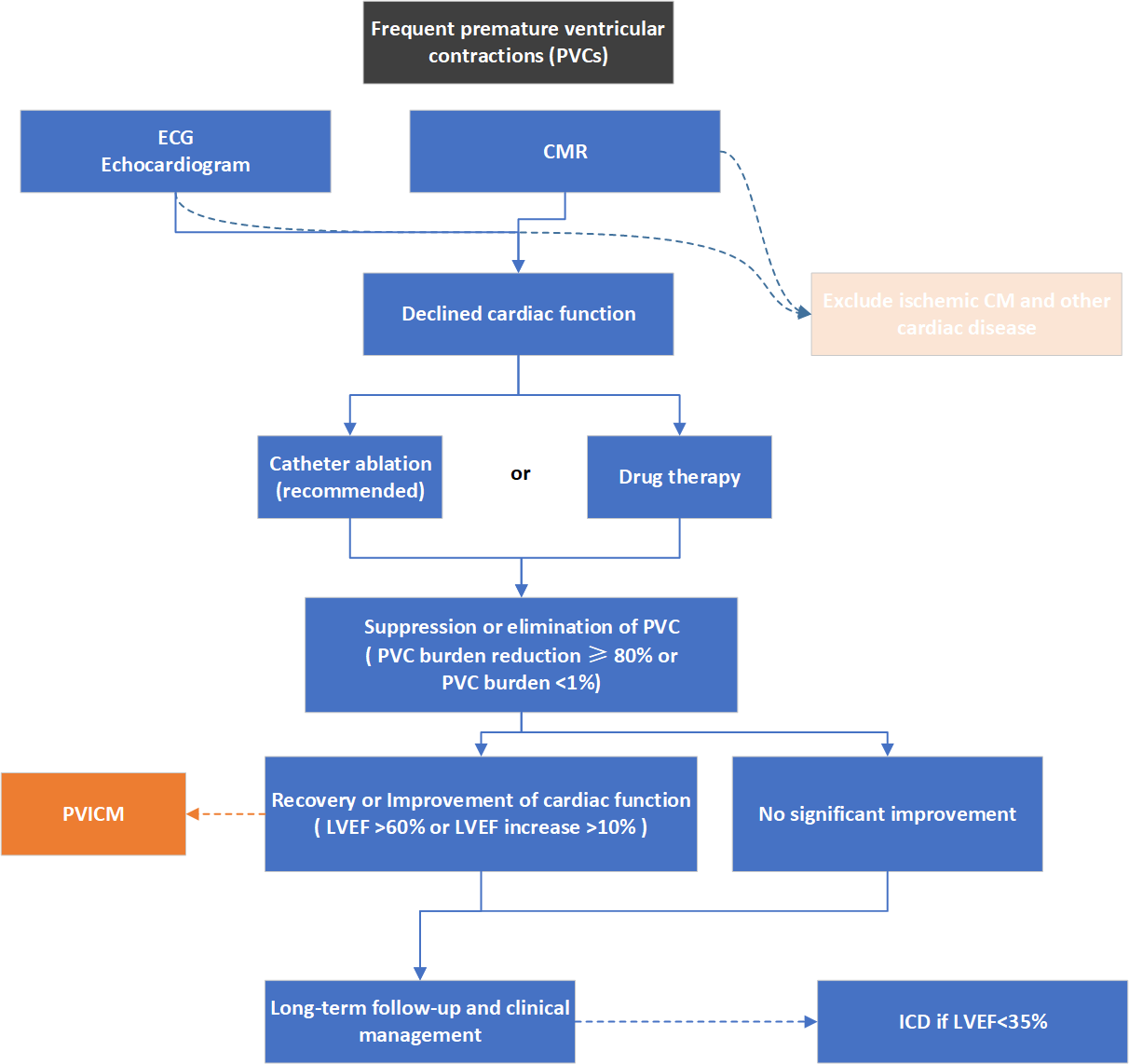 Fig. 2.
Fig. 2.Overview of a flow for diagnosis and treatment of PVICM. ECG, electrocardiogram; CM, cardiomyopathy; CMR, cardiac magnetic resonance imaging; PVC, premature ventricular contraction; PVICM, premature ventricular contraction-induced cardiomyopathy; LVEF, left ventricular ejection fraction; ICD, implantable cardioverter-defibrillator.
PVCs are common arrhythmias, and are often indicative of underlying cardiac disease. Clinical data have confirmed an emerging clinical entity of PVICM. However, many clinical studies were retrospective and non-randomized, and more prospective studies should be designed to improve the database. PVC burden is still the most robust and available risk factor for PVICM. Animal models of PVICM are still necessary to further determine the mechanism responsible for the reversible cardiomyopathy, since the association between low levels of PVC burden in humans and the development of LV dysfunction remains unclear, and awaits further investigation.
MQ provided guidance and assistance, and participated in editorial changes. XS contributed to conception and design, and drafting of the manuscript. XZ contributed to the acquisition of data, made graphs, and participated in editorial changes. LZ contributed to the conception and design, made graphs, and participated in editorial changes. XL provided supports in the conceptualization and the development of our manuscript. Furthermore, XL also provided assistance in the revision process of this manuscript. All authors gave final approval of the version to be published, and agreed to be accountable for all aspects of the review in ensuring that questions related to the accuracy or integrity of any part.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.