
- Academic Editor
Vascular calcification (VC) is a complex process of calcium deposition on the arterial wall and atherosclerotic plaques and involves interaction between vascular smooth muscle cells, inflammatory and VC mediators. The latter are independent predictors of cardiovascular morbidity and mortality and potential targets of pharmaceutical therapy. This paper is a narrative review of the complex mechanisms of VC development and in this context the potential anti-atherosclerotic effects of statins. At the initial stages of atherosclerosis VC correlates with atherosclerosis burden and in the long-term with cardiovascular morbidity and mortality. A plethora of animal and clinical studies have proposed statins as the cornerstone of primary and secondary prevention of atherosclerotic cardiovascular disease. Based on coronary computed tomography data, high doses of statins may have negligible or even positive effects on the progression of coronary artery calcification. Growing data support an increase in atherosclerotic plaque calcification in peripheral arteries (e.g., carotids), after long-term, statin-therapy. Despite the paradox of increasing VC, those effects of statins have been associated with higher plaque stability, reducing the risk of consequent adverse events. Statins seem to promote a “favorable” atherosclerotic calcification, suppressing atherosclerotic lesion expansion and their vulnerability. More studies are required to clarify the underlying mechanisms.
The term “vascular calcification” (VC) is essentially synonymous with “arterial calcification”, and describes the deposition of calcium phosphate mainly in the form of hydroxyapatite [1], and refers to two different types: atherosclerotic intimal calcification and medial calcification known as Mönckeberg’s sclerosis [2]. These two forms of calcification, which may coexist, present different localization, morphology, predisposing factors and pathophysiological effects, but both lead to increased morbidity and mortality [3].
Atherosclerotic intimal calcification, accompanied by cholesterol deposition, has been associated with atherosclerotic occlusive lesions. This process is the predominant type of calcification observed in the aorta, coronary and peripheral arteries [4, 5] and is correlated with the presence of vascular smooth muscle cells (VSMCs), as well as macrophages in lipid-rich atherosclerotic plaques [6, 7, 8]. Atherosclerotic intimal calcification is associated with classic cardiovascular risk factors like age, male sex, smoking, hypertension, dyslipidemia, diabetes mellitus as well as newer ones, such as inflammation. It is focal and the adjacent vascular wall may be free of lesions. It gradually leads to narrowing of the arterial lumen, while an imbalanced deposition of calcium in certain areas of the atherosclerotic plaques renders them vulnerable, meaning they are susceptible to rupture and consequent thrombosis. Atherosclerotic cardiovascular diseases (ASCVDs) represent the clinical manifestations of atherosclerotic calcification, such as coronary artery disease (CAD) and peripheral arterial disease (PAD), while the most common acute events of plaque destabilization are myocardial infarction and stroke. There is a complex interplay between VC, lipids metabolism and atherosclerosis progression. Statins are widely used for primary and secondary prevention of ASCVDs. Therefore, dyslipidemia alleviation by statins may become an important tool to modify VC development.
Mönckeberg’s arteriosclerosis, or Mönckeberg’s sclerosis (MScl), is a form of arteriosclerosis or vessel hardening, where calcium deposits are found in the muscular middle layer of the arterial walls (the tunica media). This condition occurs as an age-related degenerative process, but it is particularly common in diabetic and uremic patients [9, 10]. The accumulation of calcium phosphate crystals significantly contributes to vascular disease, and worsens further along with the progression of kidney dysfunction, especially in patients undergoing hemodialysis. For this reason, it has long been assumed that the medial VC results from calcium deposits and leads to vascular stiffening [2, 11, 12].
Several non-invasive imaging techniques can be employed for the screening of VC such as x-rays of the abdomen and extremities and two-dimensional ultrasound to identify the presence of calcification in carotid and femoral arteries, and the aorta [13, 14]. However, quantification of the calcification is not feasible through these techniques. Alternatively, electron beam computed tomography (EBCT) and multi-slice computed tomography (MSCT) have emerged as tools for the precise evaluation of VC [15, 16]. More recently, MSCT is capable of the accurate detection and quantification of VC using scores such as the Agatston [17] and volume score [18].
The aim of the present manuscript was a narrative literature review of the currently known pathophysiologic mechanisms of VC and most importantly to evaluate the impact of statins on them. For this purpose, we analyzed the potential mechanistic explanations of statins’ effects on intimal artery calcium deposition within atherosclerotic plaques, on the medial artery wall layer and their relationship with clinical outcomes.
A search was conducted for English language publications in MEDLINE and Embase databases from January 1990 to June 2023. The following broad search terms, including Medical Subject Headings, were used: statins, lipid-lowering, vascular calcification, intimal and medial calcification, atherosclerotic plaques, imaging techniques, calcium index, calcium score, Agatston score, echogenicity, gray-scale median (GSM) score. Except for case studies, all other types of preclinical (in vitro and animal) and clinical studies (randomized, non-randomized, prospective, retrospective) were considered eligible. The articles’ reference list was checked to identify additional relevant papers for inclusion.
The evidence behind a causal relationship between lipoproteins and bone pathologies is conflicting, but the underlying mechanisms are clearly similar which requires a deeper insight into lipid-lowering drugs, like statins, in VC.
VC resembles bone mineralization and presents into 2 types:
(1) Atherosclerotic-related calcification: this is associated with intimal artery calcification and the early stages of this process are characterized by the development of micro-calcification. Calcium phosphate hydroxyapatite crystals are deposited into the extracellular matrix of atherosclerotic lesions, and their accumulation varies between patients. Spotty distribution of bone mineralization within atherosclerotic plaques has been associated with clinical manifestations, such as acute coronary events and cardiovascular death in the long term [19]. The dynamic process of microcalcification is indicative of the risk of plaque rupture and unfavorable clinical outcomes [20]. On the other hand, macro-calcification of the atherosclerotic plaques, characterized by the deposition of large amounts of calcium, gradually decreases the lumen patency and creates a more stable phenotype [21]. This is a dual-edged sword, because heavily calcified plaques are stiff and less amenable to transcutaneous revascularization, but they appear with a low propensity to rupture. Perhaps, the inhomogeneous texture of the plaques leads to variable resistance to the hemodynamic forces in the bloodstream, which increases their vulnerability. Statins may change the localization of calcium microdeposits enhancing their deposition around the necrotic core of the atherosclerotic plaque which stabilizes the lesions [22].
(2) Medial calcification (MScl): It is usually circumferential, located in the medial layer. Most frequently is observed in diabetes mellitus and chronic kidney disease and its clinical significance is the subject of debate [9, 23]. The clinical repercussions are rare because the reduction of the lumen is minimal, unless it is overlapped by an atherogenic process, where the clinical manifestations become more evident [24, 25]. This is a bone-like morphology of the artery wall. It is believed that the lesion is produced by the fatty degeneration of the VSMCs of the middle layer, forming a mass that undergoes hyaline degeneration which then becomes calcified. MScl is not related to atherosclerosis, and its link with dyslipidemia is not established. Statins cannot alter MScl [26] and there are no current data regarding the impact of statins on arterial calcification. For that reason, our review focused primarily on the intimal artery calcification which relates to dyslipidemia and statins exert significant effects.
Inflammation plays a critical role in the advancement of atherosclerosis and
contributes to approximately 20% to 30% of the remaining risk for adverse
cardiovascular events, often linked to the rupture of unstable coronary plaques.
This relationship is supported by various studies [27, 28]. Systemic inflammatory
disorders are associated with an enhanced risk of adverse events and early ASCVDs
[29, 30]. In the early stage of atherosclerosis, inflammation is the predominant
pathophysiological mechanism that promotes plaque progression and calcification
[31]. The repeated cycles of inflammatory damage and repair ultimately lead to
calcification of the initial atherosclerotic plaques, whose progress provides an
important estimate of clinical prognosis [12]. Inflammation also plays an
important role in the calcification process, while macrophages, neutrophils, and
T cells promote extracellular matrix remodeling,osteogenic differentiation and
apoptosis of VSMCs [22, 32]. A recent experimental study showed that statin
therapy is related to increased coronary artery calcification by stimulating
inflammasome and macrophages, leading to nuclear factor-k
Until recently, it was commonly believed that VC was a passive and degenerative
process. A variety of factors are involved in the active ossification process of
the arterial wall, which either act protectively by inhibiting the calcification
of the arterial wall: fetuin-A, Matrix GLa Protein (MGP), osteopontin (OPN),
osteoprotegerin (OPG), inorganic pyrophosphate (PPi), or by precipitating calcium
and phosphorus deposition through the regulation of bone metabolism: vitamin D,
parathormone (PTH), PTH related peptide (PTHrP), Receptor Activator of Nuclear
factor
Limited data from genetic polymorphisms of some of the aforementioned factors have been found to be involved in 40-50% cases of coronary arterial calcification [46, 47]. Although the data from genetic analysis are limited, they make more robust the evidence for the contribution of bone metabolism to VC. For example, the CD73 gene deficiency generates a loss of its activity and triggers the tissue-nonspecific alkaline phosphatase, a key protein for bone formation and the main conductor of the medial VC [48].
The medial layer of the vessel wall is composed of smooth muscle cells and
elastin-rich extracellular matrix. One of the major mechanisms of arterial
calcification includes the differentiation of VSMCs into osteoblast-like cells.
This is regulated by the protein Cfba1/Runx2 (core-binding factor subunit
1
Matrix metalloproteinases (MMPs) have been long validated as important contributors of atherosclerosis development, progression, and destabilization, while preliminary data implicate their role in VC [60]. Statins may act as a stabilizing factor by inhibiting the secretion of MMPs from VSMCs and inflammatory cells, with still unknown impact on VC [61, 62].
Okuyama et al. [63] declared that despite the current belief that
cholesterol reduction with statins decreases atherosclerosis, simultaneously
statins lead to coronary artery calcification as mitochondrial toxins impair
muscle function in the heart and blood vessels through the depletion of coenzyme
Q10 and ‘heme A’, and thereby ATP generation. Statins possess a mechanism that
inhibits the synthesis of vitamin K
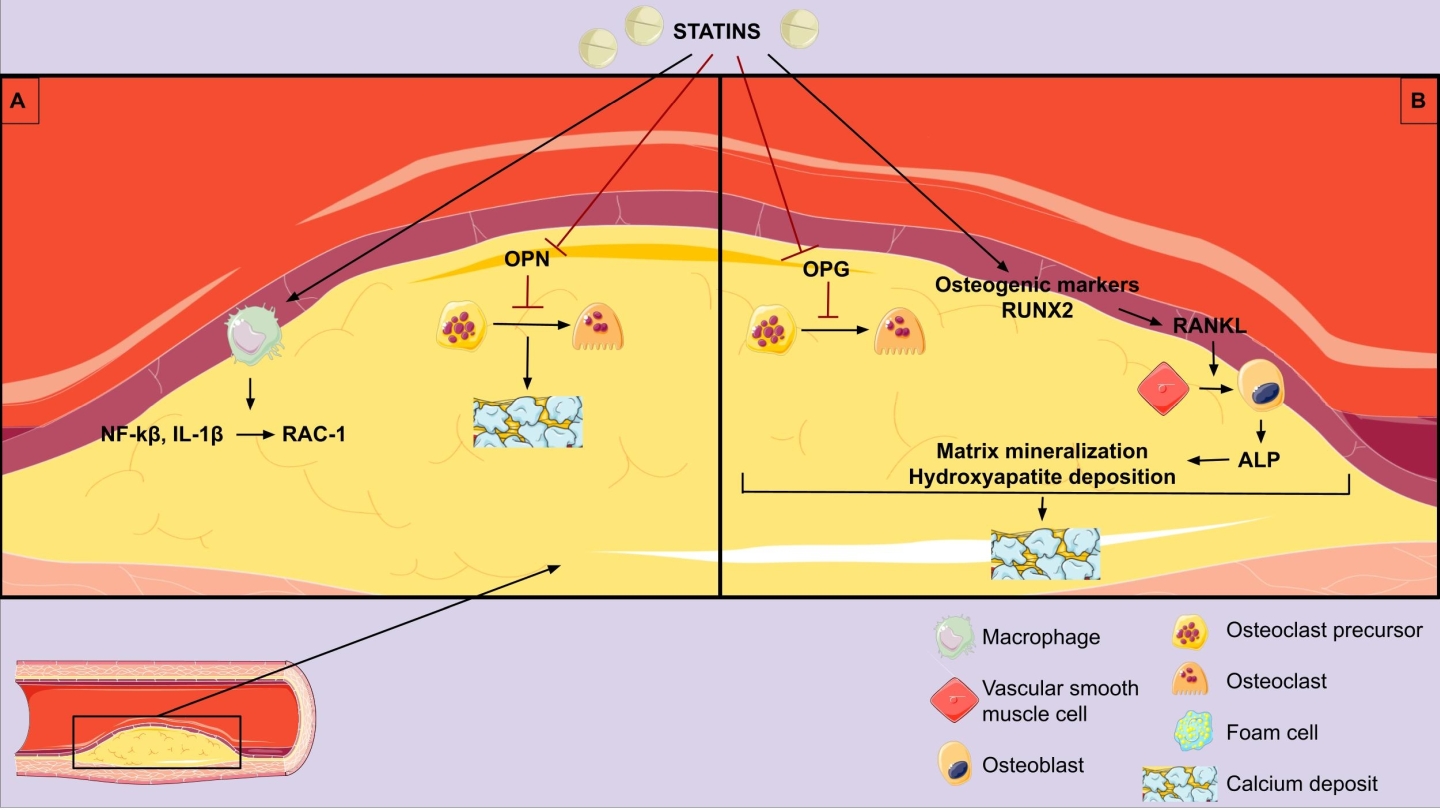 Fig. 1.
Fig. 1.Pathophysiologic mechanisms of statins on vascular
calcification. (A) Anti-inflammatory effect and vascular calcification
promotion. (B) Direct vascular calcification promotion. OPN, osteopontin; OPG,
osteoprotegerin; NF-
Standard single energy and standard dual energy chest x-rays is a low cost, low radiation diagnostic modality that can detect arterial calcifications. The chest x-ray is an already ordered procedure in almost all patients, and advanced processing has been created to enable the detection of coronary arteries calcium [64]. Also, aortic arch and peripheral artery calcification can be detected readily and reproducibly using x-rays and its presence is an independent predictor of arterial and atherosclerotic calcification, indicating CAD severity [65]. Furthermore, breast arterial calcification has been detected during mammogram screening and has been correlated with arterial calcification in the extremities and in other arterial beds, being a predictor of ASCVDs [21]. VC seen in x-rays may implicate an increased atherosclerotic calcification which cannot be distinguished from MScl.
It continues to be the most reliable and sensitive non-invasive tool for coronary and peripheral artery calcification. Since the 1940s, calcium found in the coronary arteries has served as a surrogate marker of CAD. Its presence and progression have been linked to a higher cardiovascular risk [66]. With the advances in imaging modalities, we can now detect and quantify more accurately coronary artery calcification (CAC) for cardiovascular risk assessment. For CAC quantification there are three semi-quantitative scores available: the mass equivalent score, the volume score, and the commonly utilized Agatston score. Those scoring methods exhibit a strong correlation with each other [67]. Among them, the Agatston scoring method calculates the CAC value by multiplying the area of calcified plaque by the density score. It is a widely known, valid, approach and has been endorsed by all recent international guidelines for major cardiovascular risk assessment [68]. CAC can be identified through electron beam computed tomography (EBCT) [69] and multi-slice CT [70]. These methods offer rapid scanning and processing times, taking approximately a few minutes. Additionally, the radiation dose involved is low, around 1 mSv, and there is no requirement for a contrast agent. Magnetic resonance angiography has also been employed in medical settings. However, assessment of the coronary arteries remains a challenge due to several factors such as the small size of the vessels, extended acquisition time, and the intrinsic motions caused by cardiac contractions and respiration [71]. On the other hand, CT quantification of peripheral arteries has not been clinically applicable and there is less robust evidence with scarce data. In this case, a more qualitative approach is performed in clinical practice.
It is considered a radiation free, cost effective and easily repeatable technique [72]. It is well known that there is a strong association of carotid plaque echolucency with the histologic content of vulnerable carotid plaques and the subsequent risk of a cerebrovascular event [73]. Plaque echogenicity is directly associated with the degree of calcification and fibrous tissue, and inversely associated with the lipid content of the plaque. Several scores have been proposed. In the Gray Weale-Nicolaides (GWN) classification lipid-rich plaques appear echolucent, while those with fibrous and calcific content appear echogenic [74, 75]. The Gray Scale Median (GSM) score constitutes the quantified measurement of plaque echogenicity and an important, objective, valid, marker of carotid plaque vulnerability [76] associated with increased cardiovascular mortality [77]. The GSM represents the median of the frequency distribution of tones of pixels included in the plaque areas. In other words, GSM is a median value of pixel brightness of the plaque, ignoring focal variability of the atherosclerotic lesion [76]. There are a number of factors which may influence the GSM score calculation, including the physical distance from the transducer and the consistency of the atherosclerotic plaque [73].
Also, intravascular ultrasound (IVUS) is an invasive diagnostic modality for detecting coronary calcification and stratifying plaque stability [42]. A high frequency ultrasound transducer–containing catheter is placed within the coronary arterial lumen, which detects calcium as significantly echogenic areas and provides detailed information about the distribution and nature of plaque burden. The sensitivity and specificity of IVUS detecting coronary calcification are 90% and 100%, respectively [32]. However, calcium measurement with IVUS is semi-quantitative technique limited by ultrasound’s inability to penetrate the calcium deposits [19].
Recently, there has been a significant focus on utilizing 18F-NaF positron emission tomography (PET) for the examination of VC in various arteries, including the coronary arteries, the aorta, and the carotid arteries. This technique gained considerable recognition, but its usage remains limited in clinical routine for the understanding of the underlying processes of plaque formation [78, 79].
VC is considered an actively regulated and complex process. Although highly correlated to increasing age, both types of calcification (intimal and medial) are associated with different pathological conditions such as Type 1 (T1D) and Type 2 diabetes (T2D), [80] metabolic syndrome [81], chronic kidney disease (CKD) [82], and osteoporosis affecting postmenopausal women [83]. In diabetic patients CAC has shown strong predictive value during mid and long-term follow-up compared to established risk scores, like the UK Prospective Diabetes Study (UKPDS) risk score [84]. Nevertheless, the addition of statins is not related to more rapid progression of CAC among diabetic patients [85]. Novel biomarkers have been proposed as indices of coronary calcification in CKD patients, which is of clinical importance [86]. The occurrence of arterial calcification varies significantly based on the age and gender of the individual. Research studies indicated that nearly 90% of men and 67% of women aged over 70 will develop this condition [87, 88, 89]. Factors such as increased body mass index, high blood pressure, imbalanced lipid profile (elevated LDL and TG levels), diabetes mellitus, and metabolic syndrome predispose to arterial calcification. Additionally, genetic predisposition and CKD can also contribute to arterial calcification, as identified in studies conducted by Kronmal et al. and Liu et al. [89, 90].
Following the results of the MESA study, including 6722 patients of four
different ethnic groups, Caucasian men were most likely to be identified with
higher scores of CAC
Hyperphosphatemia is highly prevalent in patients with CKD—particularly among those with advanced or end-stage renal disease—and it aggravates as the disease progresses and glomerular filtration rates decline. This impairment, especially in CKD patients undergoing hemodialysis (CKD-HD), leads to phosphorus precipitation with serum calcium and consequently to calcium phosphate deposits [94]. Increased rigidity of arterial walls and cardiac calcifications are serious complications and may increase a patient’s cardiovascular risk [95]. Arterial and atherosclerotic calcification are both very common not only in peripheral disease, but as well in coronary arteries among patients with CKD-HD [32, 42], leading to a higher risk of cardiovascular morbidity and mortality in this population [24, 25]. Among patients with advanced as well as end-stage CKD (stage 4 and 5, respectively), 50% of them have cardiovascular diseases and cardiovascular mortality accounts for ~40–50% of all deaths, compared to 26% with normal kidney function [96, 97]. Although, both atherosclerotic and medial calcifications are likely to be related to CKD-HD [98, 99, 100].
Secondary hyperparathyroidism and abnormal phosphate metabolism in CKD patients are important causes of increased VC in these patients [42]. Due to positive phosphate balance in CKD patients, this excess phosphate accumulates in the VSMCs, stimulating proteins involved in bone formation and initiating and promoting calcification [42]. Elevated CAC in these patients is associated with VSMC death in experimental models, which may lead to impaired vascular reactivity and increased plaque rupture [101]. In addition, CKD patients often have co-morbidities related to ASCVD, including albuminuria and chronic inflammation [88]. The high-risk profile of CKD patients is also associated with increased peri-intervention complications.
MScl is a type of arteriosclerosis, but it is controversial whether it extends
to the intima layer [102, 103]. Using the measurement of the ankle-branchial
index (ABI)
The interaction between atherosclerotic plaque calcification and its stability remains questionable. Scott et al. [109] have suggested the potential contribution of inflammation in promoting plaque destabilization and microcalcification. The latter represents an early, active stage of VC correlated with an inflammatory state and directly contributed to plaque rupture [110, 111, 112]. On the other hand, histological analysis of atherosclerotic plaque specimens has considered calcified plaques been more stable compared to noncalcified. There is inadequate evidence to support the CAC score as an indicator of atherosclerotic plaque stability [113].
However, statins, which are the mainstay therapy of atherosclerotic cardiovascular disease (ASVD) and reduce the risk of major cardiac events, are associated with increased coronary artery calcium (CAC) scores. Several RCTs demonstrated that statins despite their significant LDL-lowering effect, failed to reduce, but rather increased CAC scores [114, 115, 116, 117]. Statins by decreasing the soft lipid core of a calcified atherosclerotic plaque may increase the density of the plaque and its Agatston calcium score leading to smaller volume [118]. Long-term statin therapy may enhance the downstream step of calcification in the atherosclerotic process [42]. Statins’ effects in the microarchitecture of vascular calcium may be related to increased CAC and stability of the plaque [19, 119]. However, calcium density is inversely associated with event risk, suggesting that statin-induced calcification may contribute to atherosclerotic plaque stability [39, 120].
Statins, also known as 3-hydroxy-3-methylglutaryl (HMG) CoA reductase inhibitors, have been shown to reduce the risk of ASCVDs in numerous studies [121, 122]. Statins with “pleiotropic” anti-inflammatory actions effectively reduce cardiovascular events, presumably through plaque stabilization [123]. However, these drugs have also been associated with an increase in the progression of coronary and aortic calcification, which is known to be linked to an elevated risk of cardiovascular events [124, 125, 126]. Recent findings by Hanai et al. [127] suggest that lipophilic statins may have a negative impact on kidney function. The use of statins has been linked to a high CAC score, indicating a potential promotion of VC in predisposed individuals. The observed correlation between statin use and increased CAC score in the study by Li et al. [128] may be due to the phenomenon of “confounding by indication”, as statin users were older, had a higher body mass index (BMI), and had a greater burden of diabetes and cardiovascular disease. Although some studies attribute this link to confounding factors, randomized controlled trials have failed to demonstrate a survival advantage of statins in dialysis patients, leaving open the possibility that statins may not prevent or even contribute to arterial calcification. Importantly, even after adjusting for age, diabetes, BMI, and inflammation, the connection between statin use and increased CAC score persisted [129, 130, 131].
Puri et al. [126] found that although statins reduced atheroma volume, they promoted calcification in coronary atheroma. In addition to reducing the lipid-rich core of atherosclerotic plaques, statins may also increase the density of calcification [132] as part of a healing process which could result in plaque stabilization and eventually reduce the incidence of cardiovascular events. In a recent study involving 3483 participants, statin intake was linked to a 31% higher progression of coronary calcification, even after adjustment for cardiovascular risk factors [113]. Based on an old meta-analysis of controlled trials assessing the impact of statins on CAC and coronary stenoses, Henein et al. [133] concluded that the precipitated progression of coronary calcification (CAC growth rate) was due to increased transformation of noncalcified coronary atherosclerotic plaques to calcified plaques. The same authors re-analyzed data from two clinical trials to further investigate the time and dose dependent effects of statins on CAC and whether progression is accompanied by a higher incidence of cardiovascular events [112]. The included trials had the following characteristics: (1) St. Francis Heart Study (SFHS): 419 and 432 patients treated with placebo and 20 mg atorvastatin daily, respectively; CAC assessment at baseline, 2 years and 4–6 years follow-up. (2) EBEAT Study: 164 and 179 patients treated with 10 mg and 80 mg atorvastatin daily, respectively; CAC score assessment at baseline and 12 months. The accumulated data showed a similar CAC increase in the short-term follow-up (12–24 months) between placebo and low-dose atorvastatin, while 80 mg/daily atorvastatin further increased CAC by 12–14% over placebo. In the long term, a high dose of atorvastatin considerably increased the CAC score in both studies, however, that effect was not accompanied by an increase in cardiovascular events. In the SFHS trial patients experiencing cardiovascular events after the second CT scan had less-frequently prescribed statins while they had higher progression of CAC. The authors concluded that statins-induced CAC progression was not an independent predictor of cardiovascular events occurrence indicating probably plaque stabilization rather than plaque expansion [112].
In a recent multinational observational registry titled the Progression of Atherosclerotic Plaque Determined by Computed Tomographic Angiography Imaging (PARADIGM), the researchers collected data from patients who underwent serial coronary computed tomography angiography (CCTA) [134]. Statin administration actually stimulated the calcification of coronary arteries. Surprisingly, this increased calcification was associated with a reduced risk of adverse cardiac events, in agreement with previous reports [114]. In the absence of statin therapy, an increase in the CAC score indicated progression in both previously noncalcified and already calcified plaques. In contrast, the statin-related increase in CAC score indicated “calcification progression” only in previously calcified plaques [122]. In line with the findings from the PARADIGM study, Scott et al. [109] and other researchers found that statin treatment, as a primary prevention measure, correlated to a slower progression of overall coronary atherosclerosis volume, reduction of high-risk plaque features, and increased plaque calcification burden [135]. Participants with higher baseline inflammation experienced a significant increase in coronary calcification over 2 years of statin treatment, illustrating the association between inflammation, microcalcification, and the enhancing effect of statin treatment on coronary calcification. However, CAC scores did not differ between high versus low hs-CRP groups over 2 years. This further confirmed that although the CAC score is a good measure of overall plaque burden and stable end-stage macroscopic calcification, it cannot identify unstable atherosclerotic plaques [109, 112, 136]. In addition, those findings question the link of CAC score with long-term clinical outcomes [137].
Additional clinical studies shed more light on the effects of statins on plaque vulnerability. A previous meta-analysis of nine studies (a total 830 individuals) investigated the possible effect of statin therapy on the composition of coronary atherosclerotic plaques using virtual histology intravascular ultrasound (VH-IVUS) [122]. It was evident that statin administration reduced plaque volume and simultaneously increased dense calcium volume [122]. That increase has been negatively correlated with vessel remodeling in a number of studies, which may be interpreted as a stabilization of atherosclerosis [138, 139]. A recent observational study reported similar findings after the comparison of high-intensity statin treatment with standard medical treatment in patients with CAD regarding changes in plaque morphology [140]. In particular, dense calcium area increased in the high-intensity statin group compared to controls along with smoothing of atherosclerotic plaques and less shear stress and thereby less predisposition to rupture. In parallel to imaging modalities, studies assessing biomarkers have documented a positive effect of statins of VC mediators. In patients with newly diagnosed CAD, 6-months simvastatin therapy significantly reduced VC inhibitors, such as OPG, OPN and fetuin-A [141].
The possible protective effects of statins on patients with PAD has been
supported by numerous old studies. The Heart Protection Study included a large
number of participants from the UK, a subgroup of whom had PAD and were randomly
allocated to simvastatin or placebo [142]. In this sub-group of participants,
statin use was associated with a 24% lower probability of major vascular event
in comparison with the placebo group (relative risk (RR): 0.76, 95% CI: 0.72, 0.81). Another
large observation study of 155647 individuals with incident PAD investigated how
statin use may affect amputation and mortality [143]. The results indicated a
33% lower risk of amputation for high-intensity statin users in comparison to
antiplatelet-only users (HR: 0.67, 95% CI: 0.61, 0.74). A protective effect was
also observed in the low to moderate intensity statin group, but to a lesser
extent (HR: 0.81, 95% CI: 0.75, 0.86). After that robust evidence, statins’
administration is highly recommended (level of evidence: IA) in all patients with
PAD by both the American Heart Association/American College of Cardiology and the
European Society of Cardiology [144, 145]. A population-based cohort study in
Spain included 5480 adults with ABI
There are limited data regarding the effect of statin use on carotid
atherosclerosis and in particular through calcification. A number of studies have
demonstrated the stabilizing impact of statins on carotid atherosclerotic plaques
[147]. The Rotterdam study, a prospective cohort study of 1740 participants with
carotid atherosclerosis undergoing carotid MRI angiography [109]. Statin users
had a 73% higher probability of having intra-plaque calcification in comparison
to individuals who had never taken statins (OR: 1.73, 95% CI:1.22, 2.44). The
duration of statin therapy was an important factor for plaque calcification,
since only statin therapy lasting more than 48 months seemed to be associated
with calcification with a pronounced protective effect (OR: 1.74, 95% CI: 1.09,
2.77). However, three small (number of participants range: 26–33), observational
studies reported no impact of statins on plaque calcification. During 3 years
[148], 2 years [149] and 6 months follow-up [150], the administration of either
low- or high-dose statins did not significantly alter either the absolute or the
percentage of calcification within carotid plaques. However, the inability to
reach statistical significance may be due to the study design. Interestingly, the
percentage change of calcification between baseline and 6 months correlated with
percentage plaque volume change in the last study. Using ultrasound, statin
therapy may increase plaque echogenicity, GSM score and hence its stability [75].
In patients with established carotid atherosclerosis (carotid stenosis
| Reference | Participants; vascular calcification indices | Study design | Findings |
| Coronary artery disease | |||
| Schmermund A et al. (2006) [115] | 471 pts w/o CAD, w/ |
• RCT: Group A (N = 235): ATORVA 80 mg; Group B (N = 236): ATORVA 10 mg | Group A vs group B: |
| EBT: CAC | • Duration: 12 mo | ↔ CAC | |
| Kovarnik T et al. (2012) [132] | 89 pts w/ stable angina; | • RCT: Group A (N = 18): Aggressive therapy ATORVA 80 mg/d + EZET 10 mg/d; Group S (N = 71): standard statin therapy (started by GP or ATORVA 10 mg/d statin-naive patients) | Group A vs group S: |
| Coronary VH-IVUS | • Duration: 12 mo | ↑ coronary dense calcification | |
| Henein MY et al. (2011) [133] | 11 studies, 1839 pts; | • Meta-analysis | High dose vs low-dose vs placebo: |
| 6 trials assessing CAC and 5 trials assessing coronary stenoses; | • High dose statins vs low dose statins vs placebo | ↔ coronary calcification | |
| EBT, MDCT: CAC | • Duration: 12–24 mo | ↓ coronary stenosis | |
| Henein M et al. (2015) [124] | 2 clinical trials w/ 1194 pts: St. Francis Heart Study (SFHS) and EBEAT Study; | • Pooled analysis of 2 RCTs | ↑ CAC w/ greater statin doses and prolonged therapy |
| CCTA: CAC score | • SFHS study — group A (N = 432): ATORVA 20 mg/d; group B (N = 419): placebo | ||
| • EBEAT Study — group A (N = 179): ATORVA 80 mg/d; group B (N = 164): ATORVA 10 mg/d | |||
| • Duration: CAC score at baseline, 2 y, 4–6 y in SFHS study and 0 and 12 mo in EBEAT study | |||
| Banach M et al. (2015) [122] | 9 prospective clinical studies, 830 pts, 16 statin treatment arms; | • Systematic review & meta-analysis | All statins: |
| coronary VH-IVUS | • Statin intervention: 737 pts (ATORVA, 10 to 80 mg/day; PRAVA, 10 to 40 mg/day; SIMVA, 20 mg/day; ROSUVA, 10 to 40 mg/day; FLUVA, 60 mg/day; PITAVA, 2 to 4 mg/day) | ↑ coronary dense calcium volume | |
| • Placebo: 93 pts | |||
| Puri R et al. (2015) [126] | 8 RCTs, 3495 participants; | • post-hoc propensity-weighted analysis | HIST vs LIST or no statin group |
| IVUS assessment of coronary calcification and percent atheroma volume (PAV) | • HIST (N = 1545): High intensity statin therapy ATORVA 80 mg/d, ROSUVA 40 mg/d | ↓ PAV | |
| • LIST (N = 1726): Low intensity statin therapy ATORVA |
↑ coronary calcification | ||
| • No-statin therapy (N = 224) | |||
| Dykun I et al. (2016) [125] | 3483 participants; | • Observational study | ↑ CAC |
| EBT: CAC progression | • 230 pts on statins at baseline | ↓ coronary events | |
| • FU median duration: 5 y | |||
| Coronary calcification | |||
| Raggi P et al. (2005) [116] | 475 hyperlipidemic, postmenopausal women; | • RCT: Group A (N = 218): intensive statin ATORVA 80 mg; Group B (N = 257): moderate statin therapy, PRAVA 40 mg | Group A vs group B: |
| EBT: CAC | • Duration: 12 mo | ↔ CAC | |
| ↓ LDL | |||
| Houslay ES et al. (2006) [117] | 102 pts w/ calcific AS; Helical CT: CAC | • RCT: Group A (N = 48): ATORVA 80 mg; Group B (N = 54): matched placebo | Group A vs group B: |
| • Duration: median FU 24 mo | ↔ CAC | ||
| Terry JG et al. (2007) [114] | 80 pts w/ asymptomatic vascular disease, HDL |
• RCT: Group A (N = 40): SIMVA 80 mg; Group B (N = 40): placebo | Group A vs group B: |
| MDCT: CAC | • Duration: 12 mo | ↑ CAC, AAC | |
| ↓ TC, TG, LDL | |||
| Andelius L et al. (2018) [131] | 12 studies, 692 participants; | • Systematic review & meta-analysis | Intensive vs moderate statin therapy: |
| CCTA: plaque volume, plaque calcification, calcium intensity signal | • Intensive statin therapy (N = 99) | ↓ non-calcified plaque volume, | |
| • Moderate statin therapy (N = 404) | ↑ calcified plaque volume ↑calcium signal intensity | ||
| • Controls (N = 189) | |||
| • FU: 14.5 |
|||
| Lee SE et al. (2018) | 1255 pts; | • Prospective multinational registry: Group A (N = 781): statin receivers; Group B (N = 474): statin naïve | Group A vs group B: |
| PARADIGM study [135] | CCTA: CAC progression | • Duration: |
↓ atheroma volume |
| ↓ non-calcified and ↓ high risk plaques | |||
| ↑ plaque calcification | |||
| Lee SE et al. (2019) | 654 pts; | • Prospective multinational registry: Group A (N = 408): statin receivers; Group B (N = 246): non-statin | Group A vs group B: |
| PARADIGM study [134] | CCTA: CAC progression | • Duration: |
↑ calcified plaque volume |
| ↓ non-calcified plaque volume progression | |||
| Scott et al. (2022) | 142 participants; | • Prospective, cohort study, sub-analysis of RIGHT study: Group A (N = 66): high hs-CRP; Group B (N = 76): low hs-CRP | Group A vs B: |
| RIGHT study [109] | CCTA: coronary calcification | • Duration: 2 years | ↑ DCB |
| ↔ NCB | |||
| Carotid artery disease | |||
| Kadoglou NPE et al. (2008) [151] | 97 pts w/ carotid stenosis |
• Prospective study | ATORVA: |
| 52 age-& sex-matched controls; | • ATORVA (10 mg–80 mg) target LDL-C |
↑ GSM score | |
| GSM score | • Controls at baseline | ↓ OPG, OPN | |
| OPG, OPN | • Duration: 6 mo | ||
| Kadoglou NPE et al. (2010) (JVS) [73] | 140 pts w/ moderate carotid stenosis w/o indication of revascularization; | • RCT: Group A (N = 70): Low-dose ATORVA (10 mg–20 mg) target LDL-C |
High-dose vs Low-dose ATORVA: |
| GSM score | • Duration: 12 mo | ↑ GSM score | |
| OPG, OPN | ↓ OPG, OPN | ||
| Kadoglou NPE et al. (2010) (EJVS) [152] | 113 pts w/ bilateral carotid atherosclerosis; | • Group A (N = 46): Ipsilateral carotid revascularization; Group B (N = 67): Bilateral low-grade stenosis | Group A vs group B: |
| GSM score | • Both groups received ATORVA 10–80mg, LDL target |
↑ GSM score contralateral | |
| OPG, OPN | • Duration: 6 mo | ↓ OPG, OPN | |
| Mujaj B et al. (2018). The Rotterdam study [121] | 1740 pts, age |
• prospective population-based cohort study. | Higher dose and longer use of statins: |
| • statin exposure: 30.2% of participants | ↑ carotid plaque calcification | ||
| • Median duration exposure: 48 mo | |||
AAC, abdominal aortic calcium; AS, aortic stenosis; ATORVA, atorvastatin; CAC, coronary artery calcium; CCTA, coronary computed tomography angiography; CV, cardiovascular; CVD, cardiovascular disease; DCB, dense-calcified coronary burden; EBT, electron-beam tomography; EZET, ezetimibe; FLUVA, fluvastatin; FU, follow-up; GP, general practitioner; GSM, grey scale median; HDL, high-density lipoprotein; HIST, high intensity statin therapy; hs-CRP, high-sensitivity C-reactive protein; LIST, low intensity statin therapy; LDL, low density lipoprotein; LOVA, lovastatin; MDCT, multi-detector computed tomography; NCB, noncalcified coronary burden; pts, patients; PAV, percent atheroma volume; PITAVA, pitavastatin; PRAVA, pravastatin; RCT, randomized control trial; ROSUVA, rosuvastatin; SIMVA, simvastatin; TC, total cholesterol; TG, triglycerides; w/, with; w/o, without; VH-IVUS, virtual histology intravascular ultrasound; CT, computed tomography; OPN, osteopontin; OPG, osteoprotegerin.
Some studies have attempted to explain this paradoxical effect by suggesting that the statins-induced progression of calcification may occur in a way that simultaneously reduces cardiovascular risk, such as by altering the size or density of calcium deposits. For example, a reduction in mineral surface area resulting from the coalescence of small deposits and/or decreased porosity may lower the risk of debonding and subsequent plaque rupture, which could contribute to the reduction in cardiovascular risk observed with statin therapy [117, 154]. To explore this possibility, F-NaF micro-positron emission tomography (µPET) imaging, which can detect fluoride adsorption on the surfaces of actively mineralizing apatite mineral deposits, may serve as a useful tool to quantify the surface area of cardiovascular calcium deposits [155].
Several mediators have been involved in arterial and atherosclerotic calcification reflecting a complex process. VC has been associated with high cardiovascular morbidity and mortality, rendering it a potential target of therapy. Statins constitute the cornerstone of primary and secondary prevention of ASCVDs. Most studies using imaging modalities and/or biomarkers have demonstrated that statins promote atherosclerotic plaque calcification in coronary and peripheral arteries in the long term, especially at high doses. Although such an effect seems detrimental at first sight, it has been associated with higher plaque stability and less adverse cardiovascular events. Presumably, statins promote favorable arterial and atherosclerotic calcification. which do not expand atherosclerotic lesions and attenuate their vulnerability. More studies are required to verify those findings and clarify the underlying mechanisms.
MS, NV, EK and EC did the literature search, wrote the first draft of the manuscript. NV and EG created images and table. NPEK conceptualized the idea, developed the outline for the review, critically revised the manuscript and figures for submission in its final form. GV made substantial contribution to the conception and design of the work and performed the analysis and interpretation of data. GV critically revised the manuscript and figures for submission in its final form. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Nikolaos P. E. Kadoglou is serving as Guest Editor of this journal. We declare that Nikolaos P. E. Kadoglou had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Zhonghua Sun.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.