
- Academic Editor
In this comprehensive review, we examine the intricate interplay between
inflammation, ferroptosis, and atrial fibrillation (AF), highlighting their
significant roles in AF pathophysiology and pathogenesis. Augmented inflammatory
responses are pivotal to AF, potentially leading to atrial remodeling and reentry
phenomena by impacting calcium channels and atrial tissue fibrosis. A strong
correlation exists between inflammatory cytokines and AF, underscoring the
importance of inflammatory signaling pathways, such as NOD-like receptor thermal protien domain associated protein 3 (NLRP3) inflammasome,
Nuclear Factor kappa B (NF-
Atrial fibrillation (AF) is a common atrial tachyarrhythmia affecting tens of thousands globally. This atrial arrhythmia characterized by mural thrombosis and impaired cardiac function, diminishing quality of life and potentially leading to major adverse cardiovascular events (MACE) [1]. Over the past 30 years (1990–2019), the global incidence of atrial fibrillation has increased dramatically, from 2,313,549 (95% UI 1,764,441–2,950,592) in 1990 to 4,720,324 (95% UI 3,644,331–5,961,597) in 2019 [1]. Stroke, one of the most serious complications of AF, negatively affects the quality of life of AF patients, resulting in a significant burden to patients and their families. One cohort study has confirmed an overall incidence of ischemic stroke in AF patients of 30.8 per 1000 person-years during follow-up [2]. Furthermore, a clinical trial found that earlier utility of direct oral anticoagulants could reduce stroke incidence by an estimated 2.8% per month [3]. With the application of antiarrhythmic drugs, radiofrequency catheter ablation and anticoagulants, AF and its complications can be effectively controlled [2, 3]. However, the underlying mechanism for the occurrence and maintenance of AF remains unclear.
Older age, obesity, inflammation, abnormal hormone secretion, and genetic alterations are all linked to AF development [4]. At present, a growing body of evidence suggests a significant association between inflammation and AF [5]. It has been reported that ‘NOD-like receptor thermal protien domain associated protein 3’ (NLRP3) inflammasome increases AF susceptibility in obese patients [6]. NLRP3 inflammasomes in cardiomyocytes may contribute to the onset and maintenance of AF by promoting ectopic activity or reentry [4]. As a biomarker representing systemic inflammation, levels of C-reactive protein (CRP) are considered to be a prognostic factor of AF and have been positively correlated with the occurrence of AF [7]. A cohort study showed that multiple systemic inflammatory markers, including CRP, neutrophils, and macrophages, were significantly and linearly associated with AF after adjusting for statistical confounding variables [8]. Furthermore, ferroptosis, an iron-dependent form of cell death, also plays an important role in inflammatory signaling pathways [9]. Some antioxidants have shown anti-inflammatory effects as ferroptosis inhibitors in animal models [10]. Research also suggests that ferroptosis may affect tissue fibrosis through inflammation or the immune response [11]. In experimental studies, rats with chronic alcohol intake showed increased AF vulnerability; inhibiting ferroptosis reduced, suggesting a role for ferroptosis in the initiation of AF via atrial myocarditis [12]. These findings prompted our investigation into the role of ferroptosis and inflammation in the molecular mechanism for the development of AF. We sought to find a new molecular basis for the occurrence of AF, to provide a safer and more effective treatment for AF patients.
Ferroptosis is a type of cell death distinct from autophagy, apoptosis, and necroptosis, and its definition was first proposed by Brent R. Stockwell in 2012 [13]. Ferroptosis is an iron-dependent, non-apoptosis-regulated, oxidative cell death [13]. The main characteristics of ferroptosis include iron accumulation, and alterations in mitochondrial morphology, amino acid metabolism, lipid peroxidation (LPO) and other biochemical changes [13]. There are three critical events involved with ferroptosis: iron accumulation, glutathione (GSH) depletion, and LPO. These biochemical changes are the root cause of ferroptosis and other diseases. The immunological characteristics of ferroptosis are damage-associated molecular patterns (DAMPs) that release pro-inflammatory mediators such as high-mobility group protein B1 (HMGB1) [9, 13, 14]. This is one of the mechanisms by which ferroptosis causes tissues and cells to increase the inflammatory response. We summarize the characteristics and differences between ferroptosis and other types of cell death in Table 1.
| Cell death type | Characteristics of microstructure changes | Biochemical characteristics | Key regulatory factors | Immune features |
| Ferroptosis | normal nuclear; mitochondrial shrinkage; darker mitochondria; rupture of the outer mitochondrial membrane; decreasing or vanishing of mitochondrial cristae | iron overload; LPO; abnormal amino acid metabolism; reduced glutathione | glutathione peroxidase 4 (GPX4); SLC7A11; p53; acyl-CoA synthetase long-chain family member 4 (ACSL4); erastin | pro-inflammatory |
| Apoptosis | formation of apoptotic bodies; nuclear fragmentation; condensation of chromatin; plasma membrane blebbing | activation of caspases; fragmentation of oligonucleotide DNA; bare phosphatidylserine; reduced mitochondrial membrane potential | BCL2 family; caspase; BAX; BAK; p53 | anti-inflammatory |
| Autophagy | formation of autophagosomes; aggregation of double-membraned autophagic vesicles | conversion of LC3-I to LC3-II | LC3; ATG5/7; mammalian target of rapamycin (mTOR) | anti-inflammatory |
| Necroptosis | swelling of cells; condensation of chromatin; rupture of the plasma membrane | decreased ATP levels; accumulation of reactive oxygen species (ROS); phosphorylation of RIPK1/3 and MLKL; release of DAMPs | RIPK1; RIPK3; MLKL | pro-inflammatory |
| Pyroptosis | rupture of the plasma membrane; swelling of organelles; mitochondria whose integrity was not affected | activation of caspase-1; release of proinflammatory cytokines | caspase-1; gasdermins; NLRP3; GPX4 | pro-inflammatory |
LPO, lipid peroxidation; SLC7A11, solute carrier family 7 member 11; BCL2, B-cell lymphoma 2; BAX, BCL2 associated X protein; BAK, BCL2 antagonist/killer 1; LC3, microtubule associated protein light chain 3; ATG5/7, autophagy related gene 5/7; DAMPs, damage-associated molecular patterns; RIPK1, recombinant receptor interacting serine/threonine kinase 1; RIPK3, recombinant receptor interacting serine/threonine kinase 3; MLKL, mixed lineage kinase domain-like protein; NLRP3, NOD-like receptor thermal protien domain associated protein 3.
To date, ferroptosis has been shown to be involved in cell death in Alzheimer’s disease, cancer, ischemia reperfusion (including stroke and myocardial infarction), liver fibrosis, renal tubule fibrosis and other diseases [15, 16, 17, 18, 19, 20]. In view of the many characteristics of ferroptosis and its harmful effects, particularly, since the mechanism by which it caused fibrosis in cardiomyocytes is not well understood, a detailed review of the link between ferroptosis and the heart is warranted.
In general, the regulation of iron homeostasis in cardiomyocytes is similar to that in other systemic cells of the body (Fig. 1).
 Fig. 1.
Fig. 1.Metabolic Pathways associated with ferroptosis in
cardiomyocytes. Iron metabolism and cell signaling in cardiomyocytes: Nonheme
iron is transported into the cell by TF and its receptor, TFR1. Subsequently, the
endosome is acidified by ATPases, inducing the STEAP metalloreductase family to
reduce ferric to ferrous iron. Ferrous iron is released into the cytoplasm by
NRAMP2, while TF and TFR1 are transported back to the cell membrane for reuse.
Ferrous iron that is transported to the cytoplasm is oxidized to ferric iron,
which is bound to ferritin and used in enzymatic reactions or stored for later
use. Saturated ferritin is degraded by NCOA4-mediated autophagy, a process known
as ferritinophagy, and eventually, the ferrous iron produced by degradation and
the ferrous iron released from endosomes form an intracellular labile iron pool.
In GSH metabolism, the X
Physiologically, transferrin (TF) binds two molecules of ferric ions, and subsequently, they bind to the receptor of TF, transferrin receptor protein 1 (TFR1) [21]. It is this process that initiates and mediates the uptake of ferric irons by cells. In addition to this pathway, iron influx into cells can occur via the DMT-1 protein, L/T-type calcium channels at the cardiac plasma membrane, and zinc transporters [21]. Subsequently, ferric irons undergo a reduction reaction mediated by the metal reductase STEAP3 (six-transmembrane epithelial antigen of prostate) to become ferrous irons [22]. Ferrous irons are detached from transferrin in endocytic lysozymes and released into the cytoplasm by natural resistance associated macrophage protein 2 (NRAMP2), involved in the composition of labile iron pools in the cytosol [21]. Apo-transferrin and TFR1 are recycled and transported back to the cell surface.
The ferrous iron in the cytoplasm binds to the ferritin heavy chain (FTH), oxidizes to a ferric state, and forms ferritin-bound iron [22]. Iron, on the other hand, is released from the FTH by nuclear receptor coactivator 4 (NCOA4) mediated degradation of ferritin, a process known as ferritinophagy [22]. Through the above cycle, cells maintain iron homeostasis by storing excess iron ions or releasing iron ions into the cytoplasm for use when iron is needed. In addition, the excess iron can also be removed from the cell through ferroportin (FPN) [22]. In physiological conditions, labile iron is kept at very low levels, preventing the production of excessive reactive oxygen species (ROS) [21]. Nevertheless, intracellular iron overload can significantly increase the labile iron pool, resulting in the hazardous accumulation of ROS and ferroptosis.
The amino acid metabolism pathway in ferroptosis prominently features GSH, a
tripeptide composed of cysteine, glutamate, and glycine, and is ubiquitous in
cells. GSH plays a crucial role in reducing harmful per-oxidants generated by
cell metabolism to harmless lipid alcohols [23]. It exists in two forms: a
reduced form (GSH) and an oxidized form (G-S-S-G, glutathione disulfide) [23].
Extracellular cystine and intracellular glutamate enter and exit the cell in a
1:1 ratio via the cystine/glutamate reverse transporter respectively, named
system X
Phospholipids, an important component in maintaining the fluidity of the cell
membrane, is also damaged through ferroptosis [25, 26]. Polyunsaturated fatty
acids (PUFAs) in phospholipids, such as docosahexaenoic acid and arachidonic acid
(AA), are the main substances that mediate intracellular signal transduction and
are also vulnerable to peroxidation damage during ferroptosis [25]. It has been
shown that the acyl-CoA synthetase long-chain family member 4 (ACSL4) and
lysophosphatidylcholine acyltransferase 3 (LPCAT3) are involved in the
biosynthesis of polyunsaturated fatty acids in phospholipids [27].
Phosphatidylethanolamines combine with AA and adrenal acid (AdA) to synthesize
PUFAs under the catalysis of ACSL4 and CoA; and free PUAFs become bound to
membrane phospholipids through the activity of LPCAT3 [28, 29]. Acylation of
PUFAs by ACSL4, which increases the PUFA content of phospholipids, is thought to
be a specific factor for ferroptosis [27]. After synthesis, PUAFs become embedded
into the phospholipids of the cell membrane, and are subsequently oxidized by
lipoxygenases (LOXs) to become L-OOH [25]. Compounds including Fe-S and
H
The relationship between ferroptosis and fibrosis is complex. At present, it has been shown that ferroptosis can lead to tissue fibrosis in a variety of organs. A study found that the alveolar type II cells of bleomycin induced mice had iron and collagen deposition, suggesting that ferroptosis promoted the progression of pulmonary fibrosis [32]. Another study demonstrated that for simple hepatic steatosis, ferroptosis was a leading factor that led to non-alcoholic steatohepatitis, which included liver injury, infiltration of immune cells, and inflammatory cell aggregation [33], resulting in liver inflammation and fibrosis. It has also been reported that administering ferristatin-1 to mice with acetaminophen could reverse liver fibrosis [34]. However, it seems that ferroptosis is a double-edged sword for liver fibrosis. It was found that curcumol inhibited liver fibrosis by inducing ferroptosis in hepatic stellate cells [35]. It was generally believed that in parenchymal cells, ferroptosis may exacerbate tissue fibrosis; but in the case of muscle fiber cells, ferroptosis may inhibit the progression of fibrosis [29]. Therefore, there is an important link between ferroptosis and fibrosis in the process of disease formation in these organs.
A recent study revealed a notable association between cardiomyocyte fibrosis and
ferroptosis in patients with AF, with findings indicating a higher incidence of
fibrosis in this group compared to controls [36]. The specific mechanisms linking
atrial myocyte fibrosis and AF to ferroptosis, as well as the role of
inflammation in this process, remain underexplored. A study highlighted that
markers of inflammation, oxidative stress, and fibrosis were significantly
associated in AF patients [37]. Specifically, myeloperoxidase (odds ratio [OR] =
1.012, p = 0.014) and high-sensitivity CRP (OR = 1.265, p =
0.026) were independently correlated with AF compared to controls [37]. Another
experimental study also found that the administration of colchicine to
experimental rats inhibited Interleukin 1
An additional characteristic of ferroptosis is a large release of oxidized lipid mediators [39]. Undergoing ferroptosis makes cells more immunogenic, as they release DAMPs and pro-inflammatory factors that drive the tissue environment toward an inflammatory state [9, 40]. As a DAMP, HMGB1 plays a vital role in the formation of and pathological mechanism behind inflammation, as well as amplifying the inflammatory response [14]. Once released by the cell, HMBG1 gains immune-stimulating properties and acts as an adjuvant, promoting an immune response by binding to pattern recognition receptors and activating inflammation [9, 14, 40]. Furthermore, neutralizing anti-HMBG1 antibodies attenuates the macrophage inflammatory response induced by ferroptosis [41]. A possible association between ferroptosis and inflammation is shown in Fig. 2.
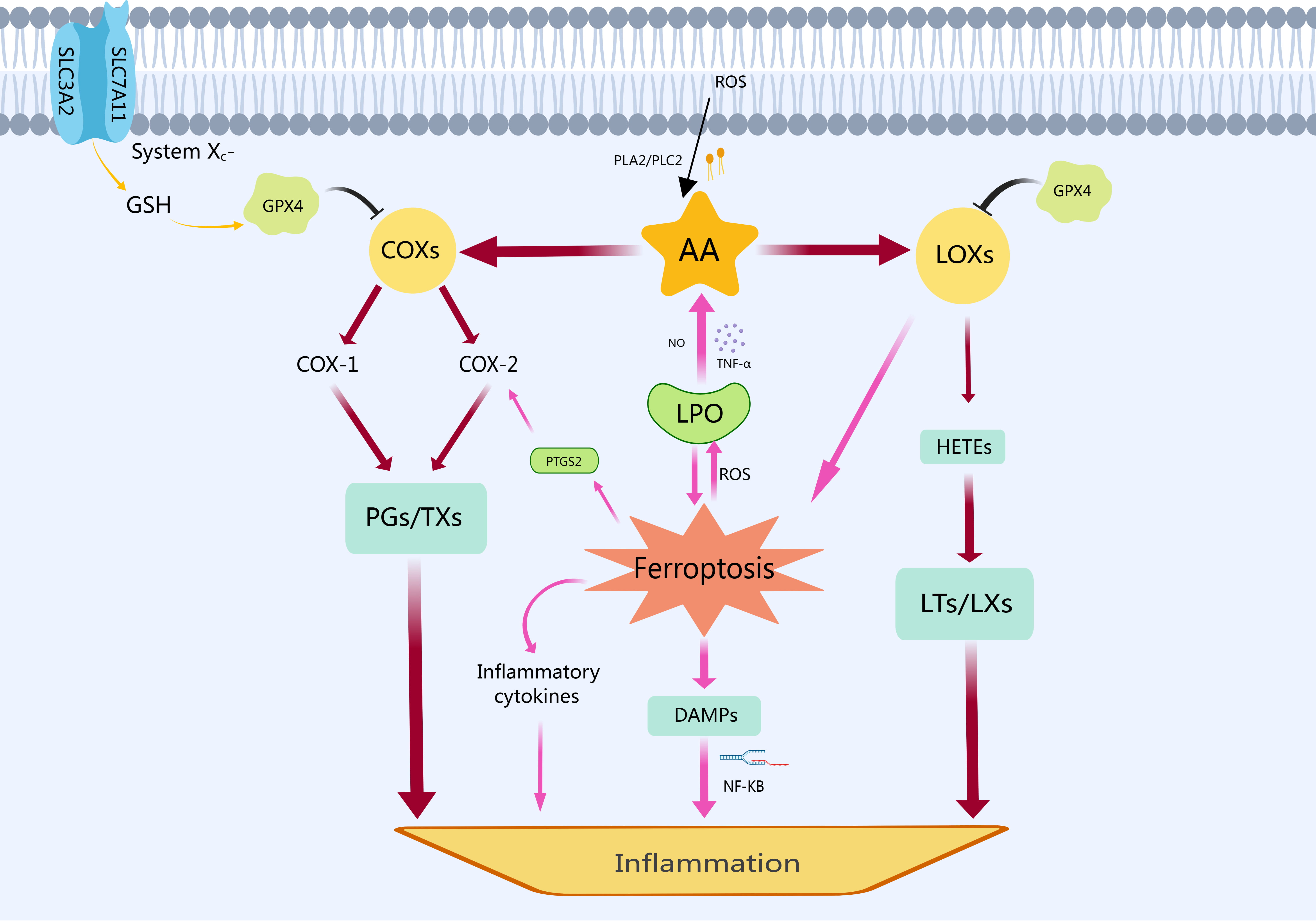 Fig. 2.
Fig. 2.The relationship between ferroptosis and inflammation. In
response to cellular stress or stimulation, phospholipase A2 (PLA2) and
phospholipase C2 (PLC2) break down cell membrane phospholipids into AA. The
polyunsaturated fatty acid AA is produced by catabolism under the stimulation of
cellular oxidative stress and LPO. Subsequently, cyclooxygenases (COXs)
metabolize AA to prostaglandins (PGs) and thromboxanes (TXs), which can induce an
inflammatory response. Simultaneously, LOXs convert AA into
hydroxyeicosatetraenoic acids (HETEs), leading to the production of leukotrienes
(LTs) and lipoxins (LXs). Additionally, LOXs can also exacerbate ferroptosis by
enhancing LPO and releasing ROS. As the core of ferroptosis, LPO perpetuates a
vicious cycle by promoting further AA decomposition through substances like
nitric oxide (NO) and tumor necrosis factor-alpha (TNF-
Previously, we discussed the metabolic pathways of ferroptosis, highlighting LPO as the underlying mechanism driving inflammation associated with ferroptosis. It is well known that PUFAs are the main components of cell membranes. PUFAs may play a crucial role in regulating inflammatory signaling and antioxidant pathways [42]. Increased intake of eicosapentaenoic acid and docosahexaenoic acid is beneficial in reducing the incidence of diseases characterized by elevated inflammation (including cardiovascular disease) [42]. PUFAs and their related metabolic enzymes have become key cellular and molecular factors resulting in inflammation [9]. PUFAs are also the most sensitive lipid in the process of ferroptosis [43, 44]. In one study, phosphatidylethanolamine (PE) was identified as a lipid associated with AF [45]. The authors suggested that PE impaired mitochondria and aggravated LPO by promoting the increase of oxidative products, triggering ferroptosis in atrial cells, and participating in atrial fibrosis [45]. Atrial cells with iron overload are in a state of cellular stress, accompanied by increased intracellular ROS, while AA is released by phospholipase C (PLC) or phospholipase A2 (PLA2) from phospholipids of the cell membrane. Additionally, AA can be oxidized by LOX, cyclooxygenase (COX) and cytochrome P450 monooxygenase, and metabolized into a variety of bioactive inflammatory mediators, such as leukotrienes (LTs), prostaglandins (PGs), and hydroxyeicosatetraenoic acid [9, 14]. These substances all contribute to the inflammatory response.
There are six LOX isoforms in the human body [43], which play an essential role in the cellular oxidation of ferroptosis. This family of lipid-peroxidases that catalyzes the peroxidation of PUFAs to produce lipid hydrogen peroxide products [43]. Furthermore LOX oxidizes AA into hydroperoxyl-intermediates including hydroperoxyeicosatetraenoic acids (HPETE) and hydroxyeicosatetraenoic acids (HETE), which are then metabolized into leukotrienes (LTs), lipotoxins (LXs) and other pro-inflammatory substances [9, 14, 44]. LOX, in addition to acting through enzyme-catalyzed lipid peroxidation, also alerts immune cells through LOX-derived proinflammatory metabolites, including LTB4/LTC4/LTD4 and LTE4, indirectly sensitizing the cell to ferroptosis [46].
COX is a key rate-limiting enzyme for the conversion of AA to PG. Two isoenzymes COX-1 and COX-2 have been found to play a key role in this process [47]. While COX-1 is widely expressed in various cell types, COX-2 is typically less expressed in normal tissues and mainly functions in activating macrophages and other inflammatory cells [47]. The gene prostaglandin endoperoxide synthase 2 (PTGS2) encodes COX enzymes, which are responsible for metabolizing AA found within cell membranes into a range of inflammatory factors including prostaglandin E2 (PGE2) [9]. Studies have shown that ferroptosis can directly increase the expression of PTGS2, and then encode more COX-2, accelerating AA metabolism, and ultimately promoting the secretion of inflammatory factors [9, 48]. Thus, inflammation induced by ferroptosis may be related to the increase of PTGS2 expression and PGE2 release.
Inflammation is also related to oxidative stress, which can trigger a range of
pro-inflammatory (such as tumor necrosis factor-
It has been suggested that inflammatory atrial cardiomyopathy, also known as
atrial myocarditis, increases AF susceptibility through tissue fibrosis,
electrophysiological remodeling, autonomic nerve remodeling, and other mechanisms
[53]. At present, a number of studies found that inhibition of NF-
 Fig. 3.
Fig. 3.The interplay between inflammation, ferroptosis, and AF.
Ferroptosis can influence atrial cells through inflammation, with atrial
inflammation being a crucial contributor to both the onset and persistence of AF.
This figure outlines a possible mechanism by which intracellular ferroptosis
induces and sustains AF through inflammatory pathways. Key factors in AF
induction include reentry and ectopic firing, which are associated with the
abnormal release of Ca
| The recruited patient group | Type of experimental tissue | Additional intervention factor | Key findings and results | References | |
| 1 | Patients with valvular disease who underwent heart valve surgery. | human tissues | —— | 1. Left atrial tissue showed significant fibrosis in the AF group compared to the sinus rhythm group. | [36] |
| 2. There was a significant increase in iron particles in the left atrial appendage tissue stained with Prussian blue in AF group. | |||||
| (Control group 13.35 | |||||
| 3. Ferroptosis was associated with atrial fibrosis in group of AF. | |||||
| (r = 0.7763, p = 0.0004) | |||||
| 2 | Patients with AF undergoing elective heart valve replacement and mice treated with PE. | human tissues and animal tissues | PE treatment group | 1. PE was a characteristic differential lipid in the AF group. | [45] |
| 2. PE promoted cardiomyocyte death by increasing mitochondrial damage and oxidative stress. | |||||
| 3. PE induced ferroptosis by inhibiting GPX4 and increasing ACSL4 expression. | |||||
| 4. Ferroptosis played an important role in atrial fibrosis induced by AngII alone and AngII combined with PE. | |||||
| 3 | Fecal microbiota from mice fed a high-fat diet was transplanted into mice fed a normal diet. | animal tissues | High-fat diet administration | 1. High-fat diet changed the composition of gut microbiota, potentially increasing susceptibility to AF through systemic inflammation. | [60] |
| 2. Pathway enrichment analysis showed that ferroptosis in high-fat diet group was significantly associated with inflammatory pathways such as NF- | |||||
| 3. The ferroptosis-related protein GPX4 was decreased and PTGS2 was increased in high-fat diet group. | |||||
| 4 | A rat model of endotoxemia established by intraperitoneal injection of LPS. | animal tissues | Sepsis | 1. The induction rate and duration of AF in the LPS group were significantly higher than in the control group. | [61] |
| 2. The expression of GPX4 was decreased and the expression of PTGS2 was increased in LPS group. | |||||
| 3. In the LPS group, total iron was increased and Fpn protein was decreased. | |||||
| 5 | Excessive ethanol-treated mouse model. | animal tissues | Excessive ethanol administration | 1. Induction and duration of AF were significantly increased in the excess ethanol treatment group. | [62] |
| 2. Atrial fibrosis was found in the excess ethanol group. | |||||
| 3. PTGS2, P53 and ACSL4 were significantly increased in the excess ethanol treatment group. | |||||
| 4. Iron accumulation was observed in the excess ethanol treated group by Prussian blue staining. | |||||
| 6 | A beagle model of AF, induced by placing a pacemaker in the carotid artery. | animal tissues | —— | 1. The transcription and translation levels of GPX4 and SLC7A11 were significantly decreased in the pacing group. | [63] |
| 2. An increase in total iron in atrial tissue was observed in the rapid pacing group. | |||||
| 3. There was a significant accumulation of malondialdehyde in atrial tissue in the pacing group. |
AF, atrial fibrillation; PE, phosphatidylethanolamine; GPX4, glutathione peroxidase 4; ACSL4, acyl-CoA synthetase long-chain family member 4; NF-
The classical transcription factor NF-
The NF-
Many studies have shown that ferroptosis is related to the NF-
The TNF-
At present, it has been concluded that TNF-
In the ferroptotic cell, when intracellular GPX4 decreases, or ROS and LPO
increases, oxidative stress will activate transcription factors such as
NF-
In the innate immune system, the NLRP3 inflammasome is the most widely studied
member of the NOD-like receptor family. The NLRP3 inflammasome is also one of the
most important components of innate immunity and plays a key role in defending
the body from pathogen invasion and the pathogenesis of various inflammatory
diseases [92]. Activation of the canonical NLRP3 inflammasome pathway consists of
two steps: priming and activation [93]. In the priming phase, which partially
coincides with the previous NF-
There is now clear evidence that the NLRP3 inflammasome plays a causal role in
the pathogenesis of AF [5, 93]. The NLRP3 inflammasome is also associated with
ferroptosis. GPX4 is an irreplaceable inhibitor of ferroptosis, and it is also
known to inhibit activation of the NLRP3 inflammasome [94]. A study has found
that a high-fat diet may induce AF through an intestinal flora imbalance leading
to lipopolysaccharide production, which could result in ferroptosis of atrial
cells and the enhancement of the TLR4/NF-
A previous study established the pathophysiological mechanism of the NLRP3
inflammasome in atrial cells in the pathogenesis of AF [95]. It was found that
the ryanodine receptor type 2 (Ryr2) Ca
Experimental studies have shown that the activation of the NLRP3 inflammasome,
when restricted to cardiac fibroblasts, can lead to atrial inflammatory changes,
atrial fibrosis, and AF [98]. Specifically, cardiac fibroblast-restricted NLRP3
inflammasome activation can intensify the activity of cardiac fibroblasts,
over-expression of connexin, atrial tissue fibrosis, and impair autonomous cell
function [98]. Pinocembrin has been reported to alleviate atrial fibrosis and
electrical remodeling by reducing the expression of NLRP3, caspase1,
IL-1
Mitochondrial dysfunction is a significant feature of ferroptosis, contributing to the occurrence and progression of AF. A recent review concluded that the effect of the NLRP3 inflammasome on ferroptosis was achieved by changing the level of ROS [100]. Similarly, the induction of the NLRP3 inflammasome by abnormal intracellular ROS levels also contributes to ferroptosis [100]. The excessive production of ROS is not only related to the inflammatory response, but is also associated with the degree of mitophagy in ferroptotic cells. Mammalian target of rapamycin (mTOR), a serine-threonine kinase, is an important regulator of cell metabolism and growth, and also participates in the regulatory mechanism of ferroptosis. Tristetraprolin and mTOR are participants in intracellular iron homeostasis, regulating iron-containing genes and the mRNA stability of the transferrin receptor 1 [101].
An experimental study has found that the ablation of the NLRP3 inflammasome can improve a series of aging-related metabolic features (including cardiac aging-related inflammation and fibrosis) [102]. This may be related to the activation of autophagy, reduction of the insulin-like growth factor 1 (IGF-1) pathway and the phosphatidyl inositol 3 kinase/protein kinase B/mammalian target of rapamycin (PIK3/AKT/mTOR) pathway [102]. Another study found that mTOR was also a profibrotic signal involved in the excessive inflammatory response and fibrotic remodeling of AF through the C-X-C chemokine ligand 12/C-X-C chemokine receptor type 4 (CXCL12/CXCR4) axis [103]. Further research also links mTOR activation with fibroblast proliferation, increased fibroblast-to-myofibroblast transformation, and cardiac collagen synthesis [104], highlighting mTOR’s involvement in AF. Over-activated mTOR stimulates the NLRP3 inflammasome, recruits pro-inflammatory factors, and contributes to AF pathogenesis by promoting fibrosis and altering atrial cell electrophysiology. Consequently, mTOR has emerged as a potential therapeutic target for AF, with its inhibition potentially reducing AF incidence.
In contrast to these favorable results, a study found that when mTOR was
inhibited in cardiomyocytes, iron accumulation occurred in the cells, leading to
iron overload [100]. This inhibition of mTOR promoted mitophagy, which often
coincides with cell autophagy, triggering the degradation of ferritin and
potentially inducing ferroptosis [100]. Sirtuin3, a classical NDA
The experimental evidence regarding the roles of mTOR and autophagy in the context of ferroptosis, inflammation, and AF is not straightforward. To address these contradictory findings, it is hypothesized that the level of mTOR inhibition and the degree of mitophagy act as a double-edged sword. Under high stress conditions, such as decreased GSH intake, decreased GPX4, and iron overload, cells experience a significant imbalance in intracellular redox metabolism, leading to intracellular LPO. At the same time, large amounts of ROS are also produced within mitochondria. Mild autophagy can be beneficial for cells, helping to remove necrotic organelles and substances exerting anti-inflammatory effects. Mitophagy, the targeted removal of damaged mitochondria, can decrease ROS production and subsequently inhibit the activation of the NLRP3 inflammasome [102].
mTOR, a critical regulator of inflammation and autophagy, exhibits contrasting
effects depending on its activity level. Overactivation of mTOR induces the
recruitment of inflammatory factors and promotes fibrosis [103, 104]. Conversely,
mild inhibition of mTOR induces mitophagy, which aids in ROS clearance and
provides cellular protection [102]. However, excessive inhibition of mTOR will
lead to severe mitophagy, which will lead to cell death, ferritin degradation and
ferroptosis [105]. Additionally, studies have also found that GPX4 overexpression
can improve mitochondrial dysfunction [107]. There was evidence that inhibition
of GPX4 resulted in LPO as well as ROS accumulation and promoted autophagy [107].
Thrombospondin 1 is thought to inhibit autophagy [107]. Overexpression of GPX4
promoted the production of thrombospondin-1 [107]. Therefore, GPX4 could block
autophagy by releasing thrombospondin 1, which eventually attenuated cardiac
fibrosis [107]. GPX4 was also thought to alleviate fibrosis by reducing
intracellular LPO and inhibiting the TGF-
In our study of the connection between inflammation and ferroptosis in AF, we
focused on the activation of the NF-
In contrast, “reactive” fibrosis can impair cardiac longitudinal conduction, but only when thicker interstitial collagen chains are present between atrial myocytes [111]. “Reparative” fibrosis, however, creates conduction blocks due to cell death and disrupted muscle bundles. Furthermore, the uneven distribution of cardiomyocytes and collagen fibers disrupts the continuity of atrial electrical signals, potentially leading to local circuit formation and promoting reentry mechanisms [111, 112]. In addition to irreversible atrial fibrosis, abnormal atrial ion channels caused by inflammation are another important mechanism leading to the occurrence of AF. It can be concluded that ferroptosis, as a type of cell death related to the heart, plays a detrimental role in inducing the inflammatory response and atrial fibrosis.
Given the central role of iron metabolism in cardiac health, we sought to identify the key molecules in the inflammatory signaling pathway associated with ferroptosis, to explore new targets for AF treatment, and to search for possible highly affinity and selective inhibitors for these targets. A study indicated that ferroptosis caused by iron overload is involved in new-onset AF in sepsis, a systemic inflammatory response, with ferroportin potentially playing a key mediating role [61]. In vitro experiments on atrial cells exposed to excessive alcohol revealed an increased susceptibility to AF, marked by elevated levels of ferroptosis-promoting molecules such as P53 and ACSL4 [62]. Icariin was shown to inhibit ferroptosis in atrial cells, reducing atrial inflammation and oxidative stress by activating the atrial SIRT1-Nrf-2-HO-1 signaling pathway [62].
Contrasting our hypothesis that ferroptosis leads to AF onset, another study proposed that AF could induce ferroptosis, especially in maintaining persistent AF [63]. This research involved rapid atrial pacing in experimental dogs, leading to the secretion of exosomes by atrial cells and cardiac fibroblasts [63]. The exosome inhibitor GW4869 reduced atrial inflammation and collagen deposition in the experimental group. In addition, they found that exosomes secreted by cardiac fibroblasts promoted ferroptosis in h9c2 cells, and that miR-23a-3p encapsulated in the exosomes may be the key molecule responsible for ferroptosis [63]. Therefore, specific inhibitors of miR-23a-3p can be regarded as a potential therapeutic target to disrupt the ferroptosis-AF cycle [63]. The above evidence strongly suggests that ferroptosis and AF are closely linked and may be complemented by atrial inflammation. These findings offer new potential targets, such as inhibiting signaling molecules or inflammatory factors in ferroptosis-related pathways. In conclusion, ferroptosis is intimately linked with AF, atrial inflammation, subsequent atrial fibrosis, and ion channel abnormalities, all of which play significant roles in this intricate process.
In this review, we discussed ferroptosis, inflammation and AF in detail, striving to elucidate the relationship between inflammation and ferroptosis. As one of the results of ferroptosis, inflammation may be involved in the pathogenesis of AF. We also reported the role of mitochondria and autophagy in the ferroptosis-AF nexus, highlighting several key enzymes and inflammatory signaling pathways. This approach offers a more comprehensive theoretical framework for understanding the association between ferroptosis and AF, especially from the perspective of ferroptosis. In addition, the detection of myocardial iron metabolism and inflammatory factors can be used as targets for the treatment of AF. At present, ferroptosis is recognized as a novel mode of cell death and has attracted considerable attention. Research in this area has predominantly focused on conditions such as cardiomyopathy, myocardial infarction, and ischemia-reperfusion. However, the exploration of ferroptosis’s connection with AF remains relatively uncharted territory. At present, it has been shown that AF is associated with ferroptosis, including the bioactive molecules related to ferroptosis [36]. Therefore, a multi-dimensional analysis of ferroptosis in the context of AF is imperative. Looking ahead, we anticipate that future experimental research will further investigate the role of inflammation in the interplay between ferroptosis and AF, and actively pursue new therapeutic targets for AF treatment.
CJ, GZ, SL contributed to the conception, design and writing of this review; CJ, ZZ contributed to the design and revision; CJ, ZZ, LG, XW, CZ contributed to the conception, revision and final review. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was funded by National Natural Science Foundation of China, grant number 82370324, and Shanghai Municipal Health Commission Health Industry Clinical Research Youth Program, grant number 20204Y0449.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.